Marfan syndrome affects the connective tissue that holds your body together. Since the syndrome affects various parts of your body, you’ll need a team of healthcare providers to manage it. Close monitoring and treatments can help you live a healthy life.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
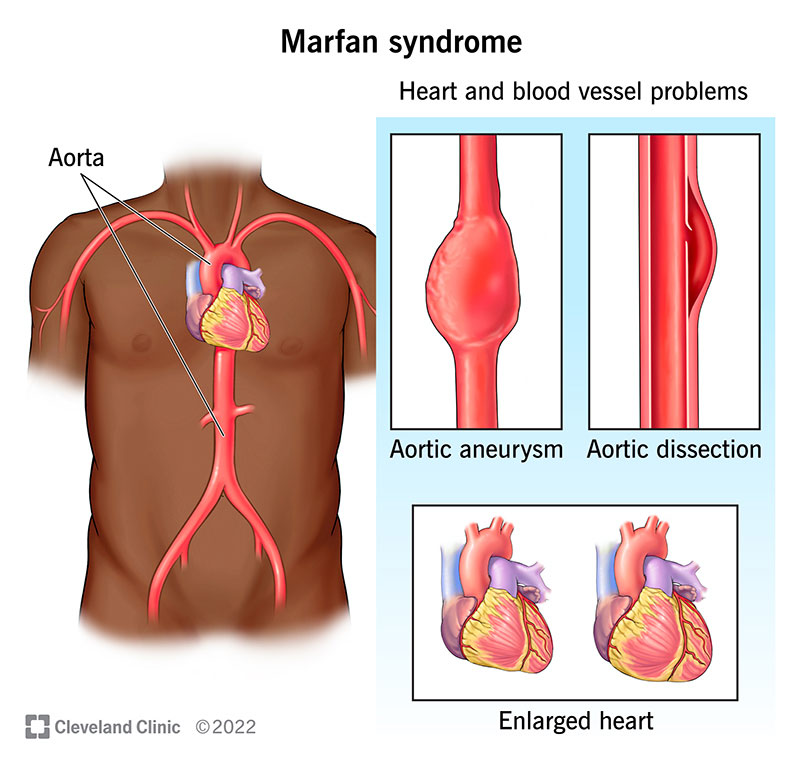
Image content: This image is available to view online.
View image online (https://my.clevelandclinic.org/-/scassets/images/org/health/articles/17209-marfan-syndrome)
Marfan syndrome (MFS) is a genetic condition that makes your connective tissue too loose and elastic. Connective tissue typically provides strength and flexibility to many structures in your body. Because of this, Marfan syndrome can affect several body systems, including your heart, blood vessels, eyes, bones and joints.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Healthcare providers call Marfan syndrome a “variable expression” genetic condition. This means not everyone has the same symptoms. The signs and symptoms vary widely in severity and when they start.
Marfan syndrome is present at birth. But you may not get a diagnosis until you’re a teen or young adult. It’s one of the most common inherited connective tissue diseases, affecting 1 in 3,000 to 5,000 people. That’s 0.02% to 0.03% of people.
The two main features of Marfan syndrome are aortic root aneurysm (widening or bulging of your aorta near your aortic valve) and dislocated eye lens (ectopia lentis).
These two issues can lead to symptoms like:
But Marfan syndrome can affect several other parts of your body, causing other symptoms. For example, physical features of Marfan syndrome may include:
Advertisement
Marfan syndrome can cause several complications affecting your heart, eyes and lungs.
Cardiovascular complications are the most common complications of Marfan syndrome (MFS). They can include:
Eye complications may include:
The changes in lung tissue that occur with MFS increase your risk for:
A genetic change (variant) causes Marfan syndrome. More specifically, there’s a change in the gene — fibrillin-1 or FBN1 — that gives your cells instructions to make fibrillin. Fibrillin is a protein that’s the main component of elastic fibers in your connective tissue.
In most cases, you inherit Marfan syndrome from a biological parent. The syndrome has an autosomal dominant inheritance. This means you only need to receive the altered gene from one parent to inherit the condition. People with Marfan syndrome have a 50% chance of passing on the disorder to each of their children.
In 25% of Marfan syndrome cases, a new gene change occurs due to an unknown cause.
Marfan syndrome can affect tissues all over your body. So, you may need a team of healthcare providers to confirm the diagnosis and develop a treatment plan.
To start, they’ll:
Healthcare providers typically use a set of criteria to diagnose MFS called the Ghent nosology. They may recommend many tests to help confirm the diagnosis or rule out other conditions, like:
A genetic test (blood test) can look for changes in FBN1, the gene that’s responsible for most cases of Marfan syndrome. But the results of genetic tests for Marfan syndrome aren’t always clear. This test can also look for other genetic conditions, like Loeys-Dietz syndrome, that cause similar symptoms.
There’s no cure for Marfan syndrome. But various treatments and strategies can help manage your symptoms and prevent complications, like:
Advertisement
You’ll need a treatment plan that’s specific to your health issues.
Certain medications can help prevent or manage complications, including:
If you get heart valve surgery for MFS, you’ll need to be on an anticoagulant medication for the rest of your life.
You’ll need routine medical appointments to monitor your:
This way, your healthcare team can track changes and catch any possible complications as soon as they appear. Your team will tell you how often you need these appointments.
Monitoring for MFS usually involves imaging tests, like:
Intense physical activity can strain your aorta and other connective tissues impacted by MFS. Because of this, you’ll work closely with a physical therapist to find exercises and sports that are safe for you.
Advertisement
Providers generally recommend low- to moderate-intensity exercise for most people with MFS. But you may need less if you have aortic root or valve replacement.
In general, you’ll likely need to avoid:
The goal of heart surgery for Marfan syndrome is to prevent your aorta from dissecting or rupturing and to treat valve problems. Together, you and your healthcare team will decide if surgery is right for you based on several factors.
The most common surgeries and procedures for MFS include:
If you need surgery, you should aim to choose a major health system that’s experienced in the type of surgery you’re getting. Your surgical team should be familiar with Marfan syndrome, as well.
If you have Marfan syndrome, you can expect a lot of medical appointments and needing to have a thorough understanding of your body. MFS affects everyone differently, so you’ll have your own journey with the syndrome. You’ll work closely with your team of healthcare providers to manage MFS as it changes.
Advertisement
Due to increased knowledge of MFS and advanced medical treatments, people with MFS live much longer than they did before the 1970s. The life expectancy of someone with MFS is now almost the same as it is for people without MFS. But life expectancy is significantly lower in males than in females.
Cardiovascular impairment is still the most common cause of death in MFS. This is mainly due to sudden death in undiagnosed cases of MFS. It’s also more likely to affect people who get a late MFS diagnosis.
Several aspects of living with Marfan syndrome can impact your mental health and quality of life, like:
Because of this, you may be at higher risk of:
Caregivers and family members of people with MFS are also at risk of these mental health issues.
Be sure to seek help from a mental health specialist (like a psychologist) if you’re experiencing distress related to MFS. Your mental health is just as important as your physical health. Joining a support group may also help.
Life with Marfan syndrome may feel like spinning through a revolving door of medical appointments, treatments and lifestyle changes. But all those check-ins can help prevent complications of Marfan syndrome so you can live the healthiest life possible. Your healthcare team will be by your side through it all. Lean on them for support and guidance. If you’re becoming overwhelmed, reach out to a mental health specialist for help.


Sign up for our Health Essentials emails for expert guidance on nutrition, fitness, sleep, skin care and more.
Learn more about the Health Library and our editorial process.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.