Lynch syndrome is a genetic condition that can lead to cancer. People with Lynch syndrome often get cancer before age 50 and should undergo lifelong cancer screenings to detect and treat cancer early.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
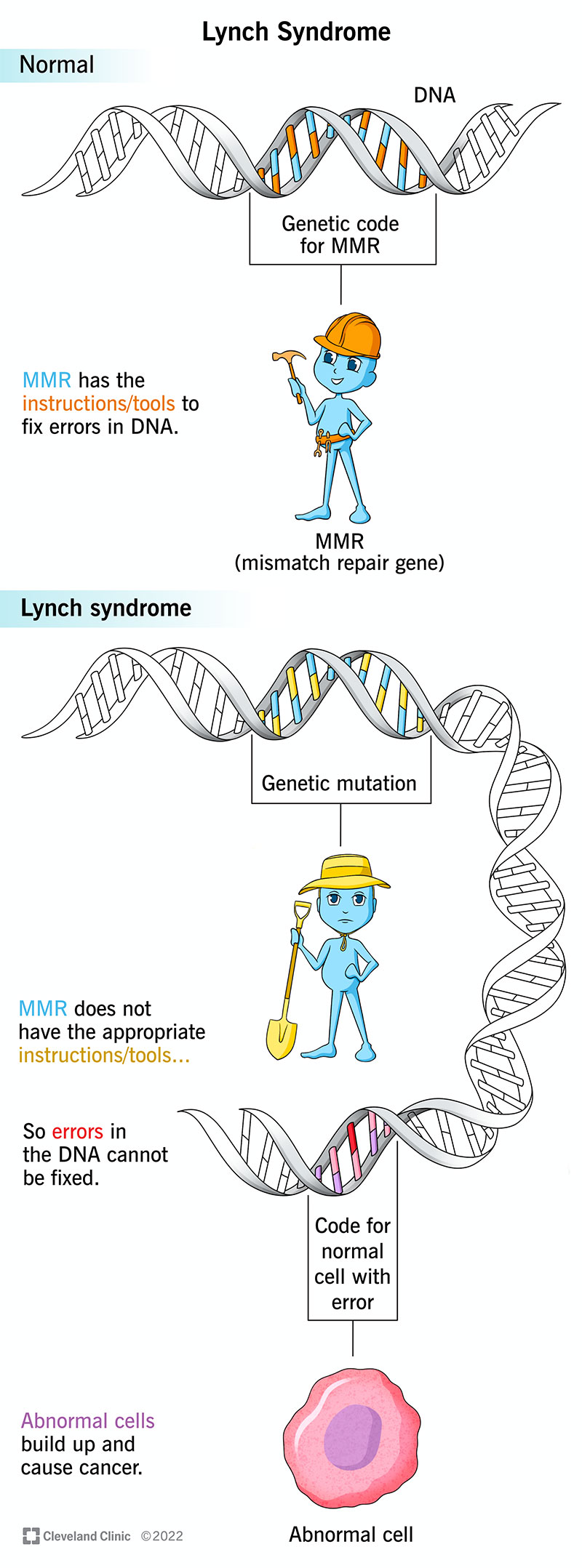
Image content: This image is available to view online.
View image online (https://my.clevelandclinic.org/-/scassets/images/org/health/articles/17195-lynch-syndrome)
Lynch syndrome is a genetic condition that increases your risk of developing cancer. People diagnosed with Lynch syndrome are more likely to get cancer before 50 years of age.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Lynch syndrome can affect anyone since it’s the result of a genetic mutation. Genetic mutations pass from your parents to you during fetal development. Sometimes, genetic mutations occur randomly, without being present in someone’s family history.
Lynch syndrome occurs in approximately one in 279 individuals in the United States. An estimated 4,000 cases of colorectal cancer and 1,800 cases of uterine (endometrial) cancer result from Lynch syndrome each year.
Symptoms of Lynch syndrome vary from person to person based on the severity of their diagnosis. People diagnosed with Lynch syndrome have symptoms similar to those of the cancers they cause, the most common being colorectal cancer.
Common symptoms of Lynch syndrome that relate to colorectal cancer include:
Not every person will experience symptoms until cancer progresses into an advanced stage. If you experience any symptoms, visit your healthcare provider.
Lynch syndrome can lead to cancer that affects several organs within your body. Types of cancer Lynch syndrome can cause include:
Advertisement
The specific organs at risk for cancer depend on which gene has a mutation in your body. The genes associated with Lynch syndrome are MLH1, MSH2, MSH6, PMS2 and EPCAM.
Colon cancers caused by Lynch syndrome tend to be more common on the right side of the colon and develop much more quickly than in the general population (one to two years versus 10 years). In addition, people who develop colorectal cancer as a result of Lynch syndrome have an increased risk of developing it again. This risk is approximately 15% within 10 years after the original surgery to remove the first cancer, 40% within 20 years and 60% after 30 years.
A genetic mutation (genetic change) in one of five genes that are responsible for fixing errors in DNA (mismatch repair gene) causes Lynch syndrome. The five genes are:
If you have Lynch syndrome, your DNA mismatch repair gene (MMR) doesn’t have all the instructions to get rid of damaged cells, so they build up in your tissues and cause cancer.
Lynch syndrome is an autosomal dominant condition. This means that only one parent needs to carry and pass the mutated gene to their child for their child to inherit the condition.
Individuals diagnosed with Lynch syndrome should tell their family members and encourage them to seek genetic counseling. Genetic counseling helps you and your family understand the risks of having a child with a genetic condition. Counseling includes an evaluation of their personal and family history as well as and genetic testing for the Lynch syndrome gene mutation.
Your healthcare provider will offer prenatal screening tests and genetic testing to diagnose Lynch syndrome before your baby is born. Genetic tests can help diagnose your child after they are born as well.
Genetic testing, which involves a blood draw or obtaining a brushing from the inside of the mouth (buccal swab), helps determine if a MLHL, MSH2, MSH6, PMS2 or EPCAM gene mutation is present in the family. If genetic testing reveals a gene mutation, your healthcare provider will confirm their diagnosis of Lynch syndrome.
If you have a Lynch syndrome diagnosis, your healthcare provider will offer regular tests to check for cancer. Tests to detect common cancers associated with Lynch syndrome include:
Advertisement
Treatment for Lynch syndrome focuses on detecting cancer and surgically removing it from your body.
Advertisement
It’s highly recommended that you receive expert care for your diagnosis. Because Lynch syndrome can affect many organ systems, the care team will include a variety of clinicians. Team members may include gastroenterologists, surgeons, gynecologic oncologists, urologists, dermatologists, gynecologists, primary care physicians, geneticists, genetic counselors and oncologists.
It’s possible that cancer could return, even after it’s surgically removed.
Some people diagnosed with Lynch syndrome choose to have a hysterectomy (surgery to remove the uterus), oophorectomy (surgery to remove both ovaries) or colectomy (bowel resection surgery) since they are at an increased risk of getting cancer in certain parts of their body.
Currently, there is no cure for Lynch syndrome. The best prognosis occurs if your healthcare provider finds and removes cancer early before it has time to spread to other parts of your body. Annual screenings, like a colonoscopy, are highly recommended for people diagnosed with Lynch syndrome.
Individuals with Lynch syndrome may develop a few colorectal polyps, called adenomas (non-cancerous tumors), in their colon or rectum. If these polyps aren’t detected and removed, they could turn into cancer. Having regular colonoscopies is important for detecting and removing these polyps.
Advertisement
You cannot prevent Lynch syndrome because it’s an inherited condition. However, people with Lynch syndrome should undergo lifelong cancer screenings, beginning in adulthood to detect cancer early.
If you have Lynch syndrome, it’s important that you visit your healthcare provider for annual checkups and work with your provider to schedule regular screening tests to detect cancer early.
Visit your healthcare provider immediately if you notice any lumps, growths or changes to your skin, which could be a sign of cancer.
Lynch syndrome and hereditary non-polyposis colorectal cancer (HNPCC) can identify the same condition, but the two conditions have a slight difference in their inheritance.
Lynch syndrome is the result of a mutation of the MMR gene. The same genetic mutation also affects people diagnosed with HNPCC, but a person’s family history of HNPCC differentiates the two conditions. Lynch syndrome can be the result of a random genetic mutation with no presence of the condition in their family history. HNPCC is always the result of a family inheritance.
No one wants to hear the phrase, “you have cancer.” With a Lynch syndrome diagnosis, that phrase might be something that you will hear from your healthcare provider, but it doesn’t have to be a negative experience. After a Lynch syndrome diagnosis, your healthcare provider will work with you to regularly schedule cancer screenings to detect and treat cancer in its earliest stages. Early detection and treatment leads to the best prognosis, so you can live a happy and healthy life.


Sign up for our Health Essentials emails for expert guidance on nutrition, fitness, sleep, skin care and more.
Learn more about the Health Library and our editorial process.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Do certain health conditions seem to run in your family? Are you ready to find out if you’re at risk? Cleveland Clinic’s genetics team can help.
