Long QT syndrome is a problem with the electrical system in your heart taking too long to recharge. This issue can lead to a life-threatening type of abnormal heart rhythm. People can inherit or acquire long QT syndrome. Most people take medication for long QT. Others may need a device or surgery to lower their risk of abnormal heart rhythms.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
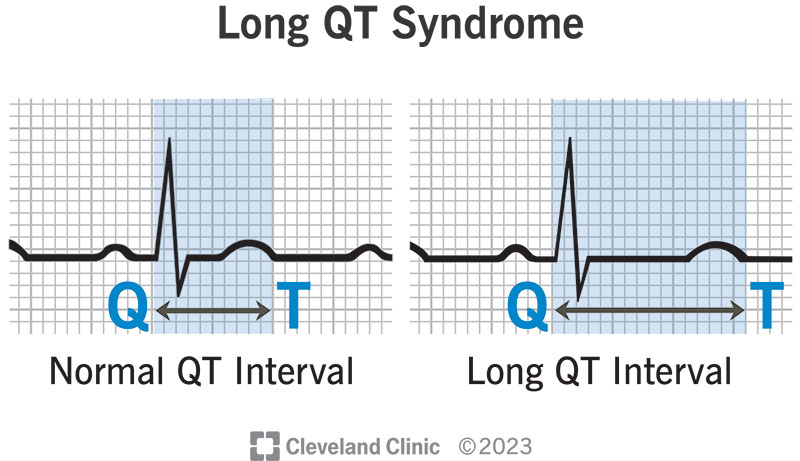
Image content: This image is available to view online.
View image online (https://my.clevelandclinic.org/-/scassets/Images/org/health/articles/17183-long-qt-syndrome)
Long QT syndrome refers to an issue with your heart’s electrical system taking longer than normal to recharge between heartbeats.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
The QT interval is the section on an electrocardiogram (ECG) report that represents the time it takes your heart muscle to contract and then recover. Put another way, it’s the time it takes for your electrical system to fire an impulse through your ventricles (lower heart chambers) and then recharge.
Ions (electrically charged particles of sodium, calcium, potassium and chloride) produce electrical activity in your heart. Long QT syndrome happens when you have an issue with the ion channels that control the flow of ions in and out of your heart muscle cells.
When these ion channels aren’t working well or you don’t have enough of them, it delays the time it takes your electrical system to recharge after each heartbeat. When the QT interval is longer than normal, it increases the risk for Torsades de Pointes, a life-threatening form of ventricular tachycardia.
Long QT syndrome has acquired and inherited forms. The types you can inherit (congenital forms) include:
Advertisement
Long QT syndrome is rare. About 1 in 2,000 people in the United States have long QT syndrome.
The most common long QT syndrome symptoms include:
The symptoms of long QT syndrome appear when the syndrome leads to Torsades de Pointes. During this arrhythmia, your heart can’t pump blood effectively. If your brain doesn’t get enough blood supply, you can faint and have a seizure. If the arrhythmia continues, sudden death will occur. If your heart rhythm returns to normal, symptoms will stop.
Symptoms are most common during:
Almost 50% of people with long QT syndrome never have symptoms. For 1 out of 10 people with this syndrome, cardiac arrest is the first sign that something is wrong. Without immediate treatment, cardiac arrest is fatal.
People who have symptoms usually start having them during their early teens. If you have symptoms of long QT syndrome, you’ll likely have them again. The risk of symptoms tends to be highest before age 30. However, people can still have them in their 50s and later.
You can inherit long QT syndrome from your parents or acquire it from certain medications. With congenital long QT syndrome, you have an abnormality in the gene code for your ion channels. Abnormal ion channels slow your heartbeat’s recovery phase. This can lead to a life-threatening arrhythmia.
People who get long QT syndrome from certain drugs are those who are inclined to have an issue with their ion channels. People with this issue may not even be aware of it until they take certain medications that trigger long QT.
Medications that can cause acquired long QT syndrome include certain drugs in these categories:
Other causes of acquired long QT syndrome include:
People at risk for long QT syndrome include:
Advertisement
Long QT syndrome is a medical condition that people can pass on from generation to generation. You should get a genetic screening if you have a first-degree relative with long QT syndrome. First-degree relatives are your parents, siblings and children.
All first-line relatives (brothers, sisters, parents and children) should have electrocardiogram (EKG) testing. Any other family members who have a history of seizures or fainting should also undergo testing.
The first step is to tell your healthcare provider that you have a family history of this condition. They may want to do diagnostic tests to check your heart. If these tests are positive, you should see a cardiologist (heart doctor) who is familiar with long QT syndrome.
Long QT syndrome can lead to Torsades de Pointes. This is a life-threatening arrhythmia that can lead to sudden death.
Healthcare providers can diagnose long QT syndrome during a routine electrocardiogram (EKG). To make a diagnosis, they measure the QT interval on the EKG. If your QT interval is longer than 450 milliseconds, you may have long QT syndrome.
Your doctor will ask you if you have a:
Advertisement
In addition to an electrocardiogram (EKG), testing may include:
Long QT syndrome treatments include medications, devices or surgery that help you manage symptoms and prevent sudden death. Treatments don’t provide a cure, but can keep you safer from abnormal heart rhythms.
Most people with long QT syndrome (even those without symptoms) take a beta-blocker such as nadolol. Other medications can shorten your QT interval or improve your electrolyte levels. Your provider will discuss which medications are best for you.
Some people with long QT syndrome also need a device, such as:
Some people with long QT syndrome may need left cardiac sympathetic denervation (LCSD) or a sympathectomy operation. This minimally invasive procedure involves removing specific nerves in your sympathetic nervous system.
Advertisement
Potential side effects or complications depend on the type of treatment you receive:
Beta-blocker
ICD
Pacemaker
LCSD
With the right treatment, people with long QT syndrome have a death rate of about 1%. Without treatment, the prognosis is poor. Up to 21% of untreated people with symptoms die within a year after they start fainting.
People with long QT syndrome can live a full life. And they can carry and give birth to children.
The inherited type of long QT syndrome is a condition people have their whole lives. However, you may have a lower risk of symptoms and complications over time.
You can’t prevent long QT syndrome if you inherited it from your parents. However, people who don’t inherit it can avoid conditions that cause the acquired type of long QT.
Long QT syndrome can affect various parts of your everyday life. You’ll need to check with your provider before doing certain things.
Startling noises or emotional distress can affect people with some types of long QT syndrome. Although you can’t control all the noises and causes of stress around you, you may be able to limit them.
You can try:
Certain types of long QT syndrome are more likely to cause issues during exercise. Your provider can give you activity guidelines based on the specific type of abnormal gene you carry. Because fatal arrhythmias can happen with exercise, you should talk to your provider when deciding to play competitive sports. You may need to take certain precautions if you decide to take part.
For some people with long QT syndrome, swimming can be dangerous. They can faint while swimming, which can lead to drowning.
Many medications can prolong the QT interval. These drugs may affect people with long QT syndrome more than people who don’t have the syndrome. If you have long QT syndrome, you should:
You should see your provider when you start having symptoms and at least once a year after that. An electrophysiologist (provider who specializes in heart rhythms) is the best option.
If your child has long QT syndrome, they should see a provider more than once a year to make sure they’re getting the right dose of medicine for their current weight.
If you have a device, you should see a provider once a year to make sure your device is working correctly.
A person who’s in cardiac arrest needs immediate medical care. A bystander should start CPR and call 911 or their local emergency number.
Questions to ask your provider may include:
Knowing you have a heart condition can make you feel anxious. Asking your provider questions and talking to a counselor can help. Be sure to keep your follow-up appointments and take your medications without missing a dose. Letting your family and friends know you have long QT syndrome can give you some peace of mind. They can call for emergency help if you start to have symptoms when they’re with you. Even better, if they know how to do CPR, they could save your life.


Sign up for our Health Essentials emails for expert guidance on nutrition, fitness, sleep, skin care and more.
Learn more about the Health Library and our editorial process.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
When your child has long QT syndrome, you’ll want care from experts familiar with this rare heart condition. Cleveland Clinic Children’s has the providers you need.
