Locations:
Glycogen storage diseases are a group of rare inherited conditions that can cause frequent low blood sugar, muscle weakness and liver damage. There are several different types based on which enzyme is missing, and each one affects you differently. Most types are manageable with treatment.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Glycogen storage diseases (GSDs) are a group of rare conditions in which your body can’t use or store glycogen properly. They’re types of inherited (passed from parent to child) metabolic disorders.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Glycogen is the stored form of glucose (sugar). Glucose is your body’s main source of energy. It comes from carbohydrates (a macronutrient) in certain foods and fluids you consume. When your body doesn’t immediately need glucose for energy, it stores glucose primarily in your skeletal muscles and liver as glycogen for later use.
Your body creates glycogen from glucose through a process called glycogenesis. When your body needs extra fuel, it breaks down glycogen for use through a process called glycogenolysis. Several enzymes are responsible for these two processes.
Glycogen storage diseases happen when you don’t have one or more of these enzymes. Your body can’t use stored glycogen for energy or maintain steady blood glucose levels. This can cause several issues, including frequent symptomatic low blood sugar (hypoglycemia), liver damage and muscle weakness.
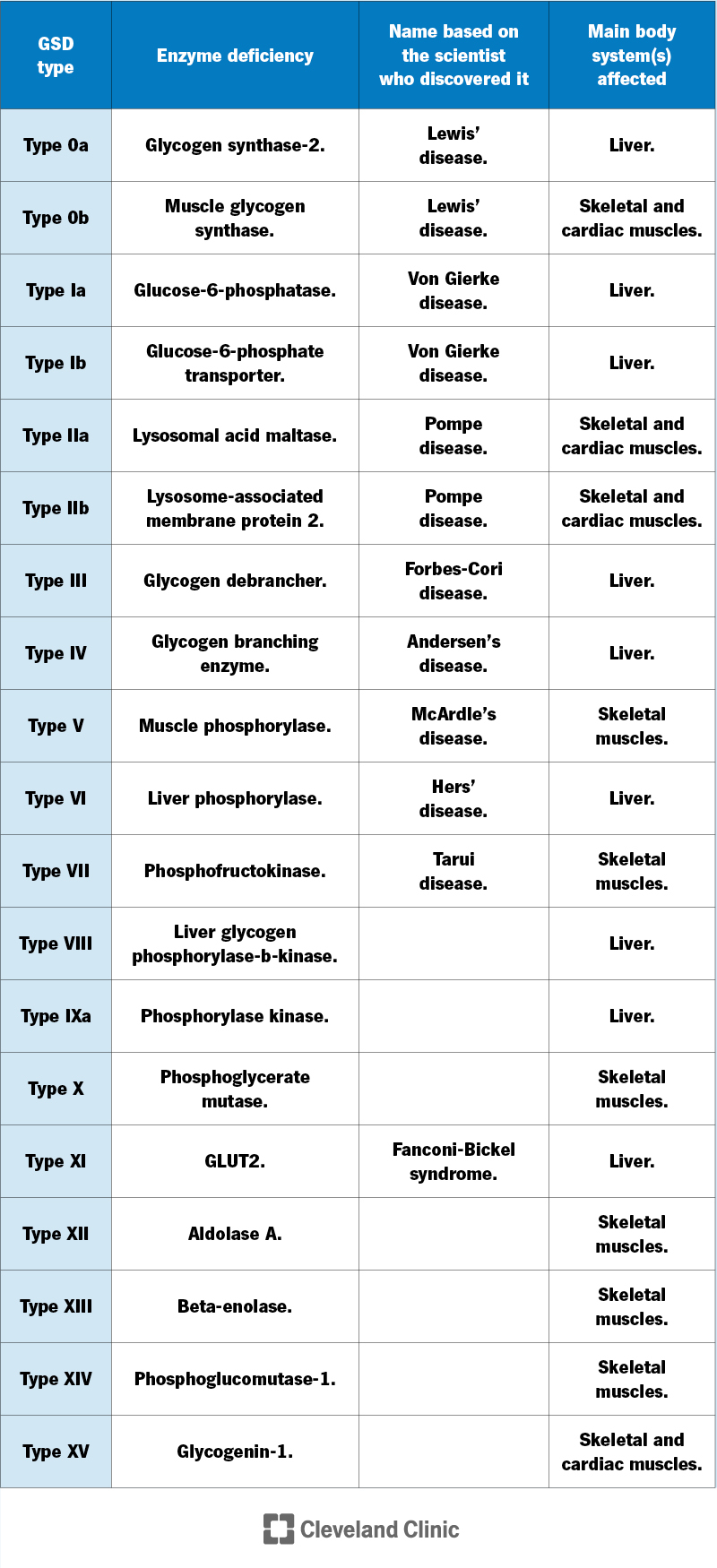
Your body uses many different enzymes to process glycogen, so there are several types of GSD — at least 19.
Researchers know more about some types than others. GSD mostly affects your liver or muscles. Some types cause problems in other areas of your body, as well.
For each type of GSD, there’s a lack (deficiency) of a certain enzyme involved in glycogen storage or breakdown. Healthcare providers may refer to each type based on the specific enzyme that’s missing or the scientist who discovered that type of GSD.
Advertisement
Glycogen storage disease is rare. GSD type I (von Gierke disease), the most common type, occurs in approximately 1 in 100,000 births.
The symptoms of glycogen storage disease can vary based on the type and even from person to person with the same type. Symptoms of GSD type 1 (the most common type) usually begin at three to four months of age. But symptoms of other types can develop later in life.
The two most common symptoms are low blood sugar (hypoglycemia) and/or getting tired easily from physical activity (exercise intolerance).
Low blood sugar happens when your blood glucose is below 70 mg/dL. Symptoms include:
Other symptoms of GSD may include:
Inherited genetic mutations (changes) that affect the function of enzymes necessary for glycogen storage and use cause GSD. A child inherits the mutations from their biological parents.
Most GSDs have autosomal recessive inheritance, meaning both parents have to pass on the mutation for the child to get the condition.
But a few types, like GSD type IX, have an X-linked inheritance. This means that your X chromosome carries the mutation. A man who has the mutation will have the condition because they carry only one X chromosome. A woman who has the mutation in one gene, with a normal gene on the other X chromosome, generally doesn’t have the condition.
To diagnose glycogen storage disease, your child’s healthcare provider will likely recommend several tests. Since GSD is rare, it can take time to rule out other conditions and figure out the specific type of GSD. The tests may include:
Advertisement
Although specific genetic testing is available for diagnosing most types of GSD, your child’s provider may want to perform a muscle or liver biopsy to confirm the diagnosis.
There’s no cure for glycogen storage disease, so treatment is based on managing symptoms. Treatment varies based on the type of GSD you have. Examples of treatment include:
Advertisement
Some GSDs (like GSD type II) can be treated with enzyme replacement therapy (ERT). This is usually an IV infusion. There’s ongoing research on using ERT with other forms of GSDs.
If you have severe liver damage from GSD, a liver transplant may become necessary.
With early diagnosis and proper management, the prognosis (outlook) for people with most GSDs is good. But some types of GSD are difficult to manage. Your healthcare provider can give you a better idea of what to expect.
GSDs can cause several complications depending on:
For example, possible complications of untreated GSD type 1 include:
The life expectancy for glycogen storage disease varies based on the type, how early it’s diagnosed and how well it’s managed. Some types are fatal in infancy while others have relatively normal life expectancies. Your healthcare provider will be able to give you the best idea of what to expect.
Advertisement
As glycogen storage diseases are genetic (inherited) conditions, there’s nothing you can do to prevent them.
If you have a biological family member with GSD, you may want to consider genetic counseling if you’re thinking of having a biological child to see if you’re a carrier of the genetic mutation.
Talk to your healthcare provider if you or your child experiences muscle weakness or persistent symptoms of low blood sugar.
Glycogen storage diseases are rare and complex. So, it’s best to find a healthcare provider who knows about the condition and how to treat it, if possible. Your child’s healthcare team will be with you every step of the way to support you and decide the best treatment for them.
Learning your child has rare glycogen storage disease can be overwhelming. Cleveland Clinic Children's experts are here to help treat and manage your child’s GSD.

Last reviewed on 06/26/2023.
Learn more about the Health Library and our editorial process.