A Chiari malformation is a structural abnormality in your skull that causes part of your brain to move into your spinal canal. You may have mild or severe symptoms or no symptoms at all. It usually leads to headaches and difficulty with balance and coordination. Surgery may help.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
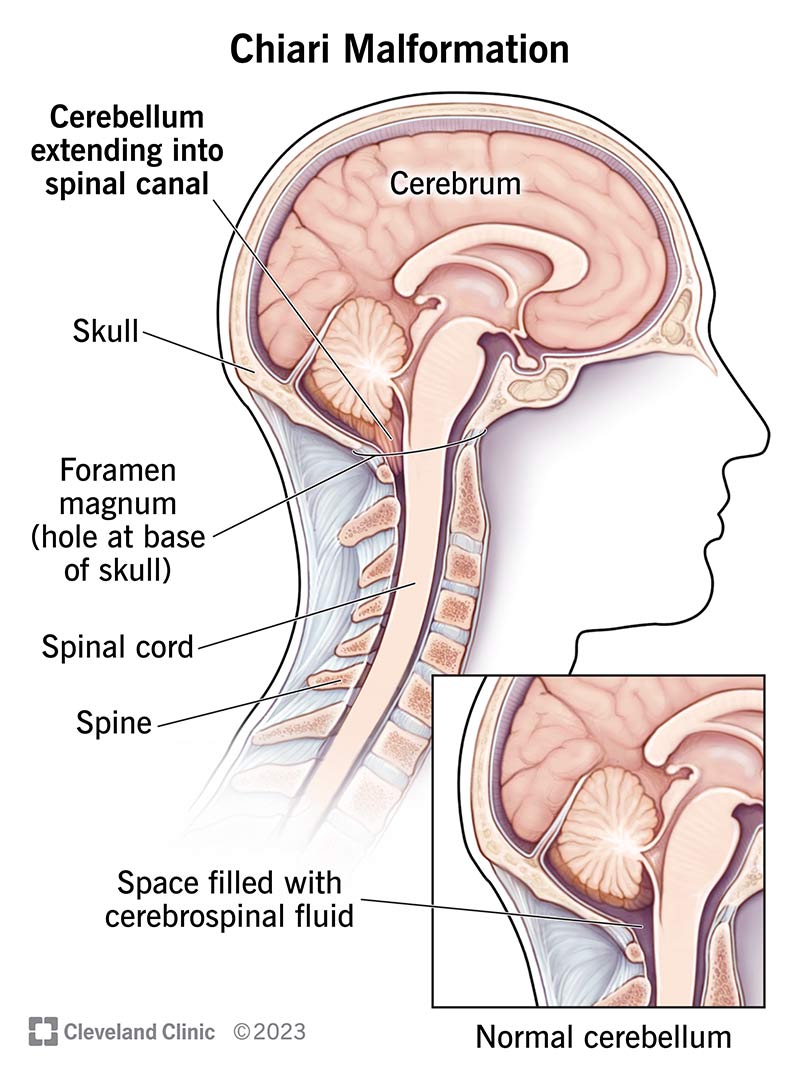
Image content: This image is available to view online.
View image online (https://my.clevelandclinic.org/-/scassets/images/org/health/articles/6008-chiari-malformation)
A Chiari malformation (CM) happens when brain tissue in the lower back of your skull goes into your spinal canal. A structural problem with your skull typically causes it. Nearly all CM cases are present at birth (congenital). But you might not know you have it until later in life.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
With CM, the base of your brain — your cerebellum — goes downward into an opening in your skull called the foramen magnum. This opening sits above your spine and spinal cord. CM can lead to pressure on your cerebellum and other nerve structures. It can also block the flow of cerebrospinal fluid (CSF). CSF helps protect your brain and spinal cord.
Healthcare providers classify Chiari malformations into types based on how much they affect the back of your brain. The types increase in severity and include:
Advertisement
Your cerebellum helps with functions like movement, balance and breathing. CMs cause symptoms related to these functions. Treatment depends on the severity and may include surgery.
You may have no symptoms with CM. And specific Chiari malformation symptoms vary based on the type. But in general, they include:
These issues can affect your mood. You may be at a higher risk of depression.
CM type 0 is rare. Symptoms may happen if a fluid-filled cyst (syrinx) forms inside your spinal cord. You may have:
This is the most common type. You may not know you have CM type 1 until adolescence or adulthood. Many people don’t have symptoms. But some people develop:
Symptoms may look different in infants and children. They may include:
CM type 2 almost always happens alongside myelomeningocele. This occurs when your baby’s backbone and spinal canal don’t close before birth. You may get this diagnosis during pregnancy and/or your baby’s healthcare provider may notice symptoms shortly after birth.
This type is associated with other conditions and complications, like:
CM type 2 causes a range of symptoms due to these related complications. They may include:
CM type 3 is very rare. It involves an encephalocele — a condition where brain tissue grows through an opening in a newborn’s skull. The symptoms and complications can range in severity. Severe cases often lead to death. Symptoms may include:
Advertisement
Differences in skull structure often cause Chiari malformations. These differences typically happen during fetal development due to a gene change. You may inherit the gene change from your biological parent(s). Or it may happen randomly after conception. There’s nothing you can do to prevent Chiari malformation. It can affect anyone.
This condition almost always develops during fetal development. So, it’s present at birth (congenital). But you may not develop symptoms until later in life.
Very rarely, Chiari malformations can develop at some point after birth. In these cases, your skull might change shape due to:
Chiari malformation can cause severe health issues and developmental delays. Complications and related conditions may include:
Advertisement
In addition, symptoms of a Chiari malformation can affect your mood, especially if you have insomnia or severe headaches. Some people may develop depression. If this condition affects your mood, talk to your healthcare provider or a mental health professional.
Imaging tests of your brain are the main way healthcare providers diagnose Chiari malformations. But to start, your healthcare provider will ask about your symptoms. They’ll do a physical and neurological exam.
Imaging tests may include:
A provider may spot a CM with imaging tests you get for other reasons.
In some cases, a provider may see a Chiari malformation in a fetus during a pregnancy ultrasound.
Your healthcare provider will make a treatment plan based on your symptoms. The goal of treatment is to support normal CSF flow and protect your brain and spinal cord from further damage.
If you don’t have symptoms, you likely don’t need treatment. Instead, your provider will recommend regular imaging tests to check whether it’s getting worse.
Treatment for mild symptoms may include:
Advertisement
Severe Chiari malformation cases may need surgery. In general, surgery helps relieve pressure on your brain and/or improve CSF flow. Procedures may include:
Each of these procedures has potential risks and complications. Your care team will explain them.
The outlook varies depending on the severity and type of Chiari malformation. CM type 1 often has a good outcome. Surgery helps improve symptoms in many cases.
Chiari type 2 and 3 can range in severity. A common complication of these types is hydrocephalus, a buildup of cerebrospinal fluid. It can be life-threatening.
It’s important to work closely with your care team. They can help monitor your symptoms and determine the right treatment plan for you or your child.
If you have mild or no symptoms after a Chiari malformation diagnosis, you’ll likely have a normal life expectancy. Severe symptoms and certain types of Chiari malformation can be fatal. Talk to your healthcare provider about what to expect after a diagnosis.
You may have a lot of questions after a Chiari malformation (CM) diagnosis. Due to the different types and range of severity, it affects each person differently. That’s why it’s key to lean on your care team for support and answers. They can give you a better idea of what to expect based on your unique case. They’ll guide you through treatment options and how this condition may affect you.


Sign up for our Health Essentials emails for expert guidance on nutrition, fitness, sleep, skin care and more.
Learn more about the Health Library and our editorial process.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Learning you may have Chiari malformation can leave you wondering what’s next. Cleveland Clinic’s expert team is here to help you get the care you need.
