Cystic fibrosis (CF) is a genetic disease that causes sticky, thick mucus to build up in your body. This can damage your lungs, pancreas and other organs. It can be difficult to breathe and to get enough nutrition. People with CF often get frequent infections. Management includes methods of clearing your airways, medications and a special diet.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
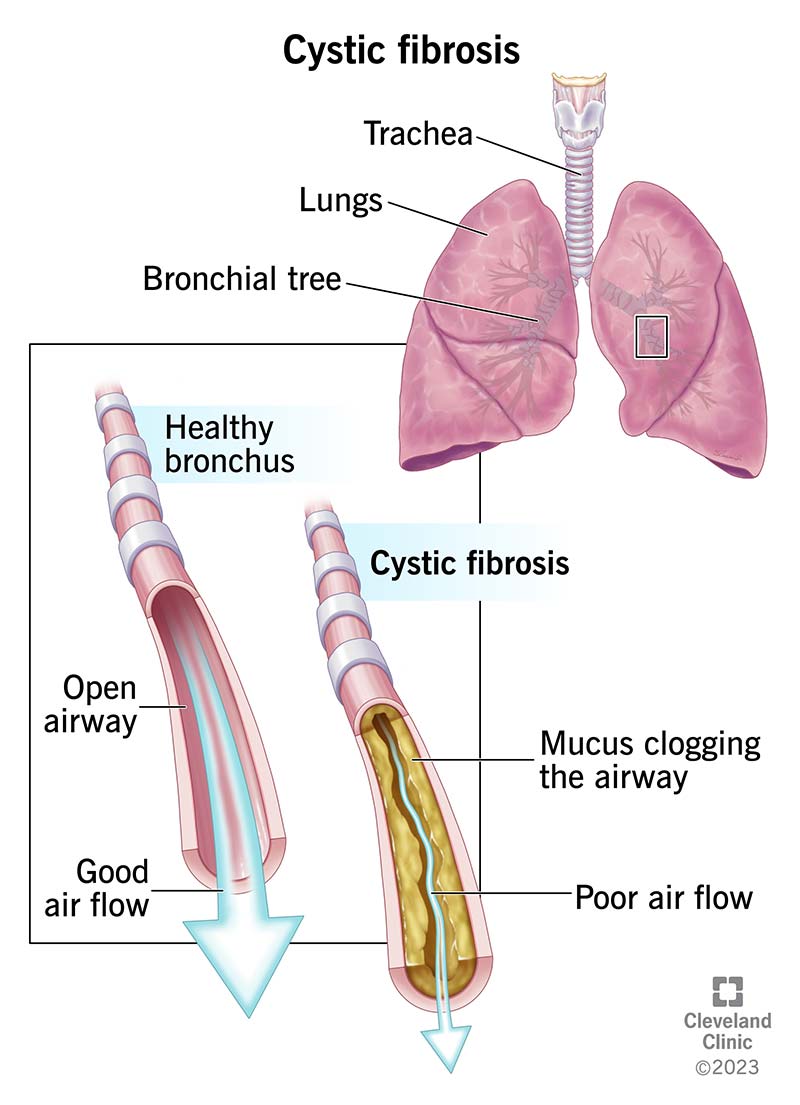
Image content: This image is available to view online.
View image online (https://my.clevelandclinic.org/-/scassets/images/org/health/articles/cystic-fibrosis)
Cystic fibrosis (CF) is a genetic disease that causes sticky, thick mucus to build up in your organs, blocking and damaging them. Many people think of CF as a lung disease because it affects your lungs and airways, which can make it hard to breathe and cause frequent infections. But it’s called cystic fibrosis because it also causes cysts and scarring (fibrosis) in your pancreas. This damage, plus the thick mucus, can block ducts that release digestive enzymes, making it hard to get nutrients from your digestive tract. CF can also affect your liver, sinuses, intestines and sex organs.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
The mucus that lines your organs and body cavities, such as your lungs and nose, is thin and watery. In people with CF, a change in a gene (genetic mutation) leads to low levels of certain proteins, or proteins that don’t work properly. Because of these faulty proteins, minerals that move water into your mucus (which thins it out) get trapped inside cells, leaving the mucus thick and sticky.
People with cystic fibrosis are born with it. It’s a lifelong illness that gets more severe over time. Most people with CF don’t live as long as people without it.
There are two types of cystic fibrosis:
Cystic fibrosis symptoms include:
Advertisement
People with atypical cystic fibrosis may have some of the same symptoms as those with classic CF. Over time, you also might experience:
Changes to the CFTR gene — called variants or mutations — cause cystic fibrosis. CFTR makes a protein that works as an ion channel on the surface of a cell. Ion channels are like gates in a cell’s membrane that allow certain molecules to pass through.
CFTR usually makes a gate for chloride ions, a type of mineral with a negative electrical charge. Chloride moves out of the cell, taking water with it, which thins out mucus and makes it more slippery. In people with CF, gene mutations in CFTR prevent this from happening, so the mucus stays sticky and thick.
There are different categories (classes I to VI) of gene mutation in CFTR that depend on the effect they have. Some produce no proteins at all, some produce only small amounts of proteins, and some produce proteins that don’t work properly.
Yes, cystic fibrosis is a genetic condition that you’re born with. People who have CF inherit two mutated CFTR genes, one from each biological parent (it’s inherited in an autosomal recessive manner).
Your parents don’t have to have cystic fibrosis for you to have CF. In fact, many families don’t have a family history of CF. Someone with just one copy of the gene variant is called a carrier. About 1 in 31 people in the U.S. are carriers who have no CF symptoms.
You’re born with the mutation in the gene that causes cystic fibrosis. But with mild symptoms, or symptoms that come and go, some people may go undiagnosed until later in life, even as adults.
Complications of CF include:
Advertisement
Healthcare providers often test for cystic fibrosis during a newborn screening. Providers perform this test with a few drops of blood from your baby’s heel. A lab looks in the blood sample for immunoreactive trypsinogen (IRT), a chemical made in your pancreas. People with CF have higher levels of IRT in their blood. Babies are often tested for IRT shortly after birth and a few weeks later.
Some conditions — like preterm delivery — can raise IRT levels. So, a positive IRT test alone doesn’t mean your baby has CF. If your baby has higher levels of IRT than expected, your healthcare provider will order additional tests to make a final diagnosis.
In about 5% of cases, the newborn screen doesn’t detect elevated IRT levels in someone with CF. Or you may have been born before routine CF screening was available. If you or your child has symptoms of CF, a provider will perform a sweat test and follow up with additional tests as needed.
Advertisement
There’s no cure for cystic fibrosis. You can manage the disease and its symptoms with the help of a cystic fibrosis specialist and other providers on your healthcare team. Management involves:
You can help to keep your airways clear if you have cystic fibrosis in a number of ways:
Advertisement
CFTR modulators are medications that can help correct issues with proteins made by mutated CFTR genes and increase the amount of functioning proteins on your cells’ surfaces. They’re not a cure for CF. But for certain people, they’ve made dramatic improvements in symptoms and life expectancy. Despite this, some people with CF don’t qualify for or can’t tolerate modulator therapies.
CFTR modulators include:
Your provider may also prescribe medications that reduce inflammation, treat infections or manage symptoms. These include:
If you have CF, your dietary needs are different from those of someone without CF. CF can prevent your pancreas from creating or secreting enzymes that help break down food. This means your intestines don’t fully absorb nutrients and fats from foods.
Your CF specialist or a registered dietitian may recommend a nutrition plan. It could include:
You may need surgery for cystic fibrosis or one of its complications. These might include:
Yes, cystic fibrosis can be life-threatening. Lung damage — from thick mucus and frequent lung infections — is the most common cause of death.
Experts predict the life expectancy of someone born with cystic fibrosis in the past few years is around 50 years old. Improvements in treatment in recent years have increased this from a few years ago, when life expectancy was between 30 and 40 years old.
People with atypical cystic fibrosis tend to have longer life expectancies than those with classic CF.
There’s no cure for CF. You or your child will need lifelong treatments to manage it. This includes treating infections, maintaining nutrition and seeing a CF specialist frequently. But new treatment methods help children who have CF live well into adulthood and have a better quality of life.
Treatments work best when CF is diagnosed early, which is why newborn screening is so important. The addition of CFTR modulators at a young age may improve long-term health and increase life expectancy even more in the future.
Since you’re born with CF, there’s no way to prevent it. If you’re a carrier of a CFTR gene variant, you can ask your provider about prenatal genetic testing and the chances that your biological children would have CF.
Taking care of yourself with CF includes developing a treatment plan with your healthcare team. You must follow this plan very closely to stay well, including:
Get recommendations from your providers about a healthy eating plan and physical activities that are safe for you. Ask your provider if pulmonary rehabilitation is a good idea for you.
You can reduce your risk of infections by avoiding people who are sick, practicing good handwashing techniques, and getting any recommended vaccinations.
You can also take part in clinical trials, which test new treatments for CF. Ask your provider if any would be a good fit for you. Make sure you get all the information about the benefits and risks of clinical trials.
Keep all of your scheduled appointments with members of your healthcare team. Talk to your provider if you have any concerns about your treatment plan or symptoms you’re having. Ask them what to do if you have symptoms of an infection. You can also reach out to them if you need help with social or emotional issues.
Go to the emergency room if you have symptoms of severe illness, including:
You might want to ask your healthcare provider:
Healthcare professionals usually recommend that people with cystic fibrosis aren’t in close contact with one another. This is because people with CF are more likely to get infections that other people fight off easily. They’re more likely to pass the germs on to others with CF (who also can’t fight them off easily). People with CF also should avoid anyone who’s sick.
It can be daunting to be diagnosed with an illness that’ll require lifelong management. But new treatments and a better understanding of cystic fibrosis give you time to take things day by day. Most people with CF are now expected to live full lives into adulthood. And it’s likely that a child diagnosed with CF today will have even more treatment options in their future.
Gather a trusted team of loved ones and medical professionals that can help you understand what to expect, and to navigate concerns that come up in daily life. And don’t be afraid to seek out second opinions anywhere along the way.


Sign up for our Health Essentials emails for expert guidance on nutrition, fitness, sleep, skin care and more.
Learn more about the Health Library and our editorial process.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cystic fibrosis can affect every aspect of your life. Cleveland Clinic’s providers can treat and help you manage CF as you plan for the future.
