A pheochromocytoma is a rare but treatable tumor that forms in the middle of your adrenal gland. In most cases, the tumor is benign, but it can be malignant (cancer). Symptoms include high blood pressure and headaches, though you may not experience any symptoms.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
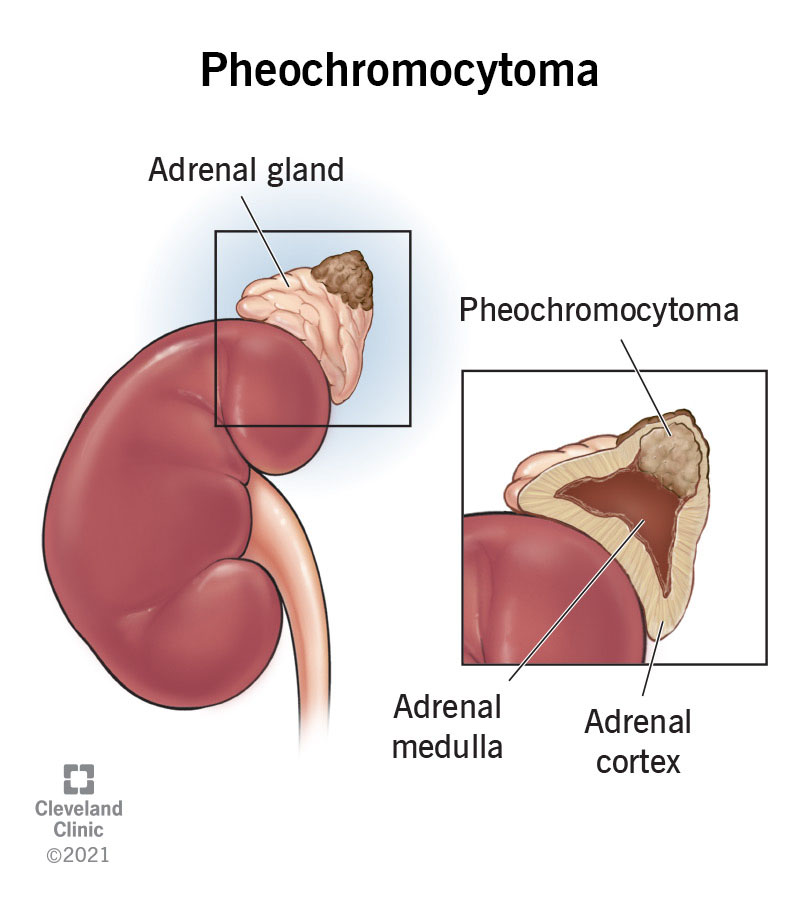
Image content: This image is available to view online.
View image online (https://my.clevelandclinic.org/-/scassets/images/org/health/articles/23373-pheochromocytoma)
A pheochromocytoma (pronounced “FEE-oh-KROH-moh-sy-TOH-muh”) is a rare tumor that forms in the center of your adrenal gland(s). The tumor releases hormones that cause a “fight-or-flight” response, leading to high blood pressure, sweating and other symptoms.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Usually, a pheochromocytoma affects only one adrenal gland, but it can also affect both. Sometimes, there’s more than one tumor. People between the ages of 30 and 50 are most likely to get one. The true number of pheochromocytoma cases is unknown because many people don’t have symptoms. But experts know it’s rare.
Most pheochromocytomas are benign (not cancerous). Approximately 1 in 10 cases may be malignant (cancerous).
All pheochromocytomas have the potential to spread to other parts of your body. Because of this, lifelong follow-up is recommended. A pheochromocytoma may be described as:
Pheochromocytoma symptoms happen when the tumor releases too much adrenaline (epinephrine) or noradrenaline (norepinephrine) into your blood. But some pheochromocytoma tumors don’t cause symptoms.
Common symptoms of pheochromocytoma include:
Advertisement
You may hear these symptoms called the five Ps of pheochromocytoma: pressure, pain, perspiration, palpitations and pallor.
Less common symptoms include:
If you have pheochromocytoma, you may have symptoms after certain events, like:
Symptoms of pheochromocytoma can be constant or come and go. Some people have ongoing symptoms, while others have sudden episodes that happen occasionally.
In most cases, pheochromocytoma happens randomly, and the exact cause isn’t known.
About 7 in 20 people who have pheochromocytoma have a genetic condition (passed through family) that’s linked to it. They include:
Talk to a healthcare provider if you have any biological first-degree relatives (siblings and parents) who’ve been diagnosed with pheochromocytoma or have any of these genetic conditions. Your provider can run genetic tests to see if you’re at increased risk.
Without treatment, pheochromocytomas can potentially cause serious complications, including:
It may also increase your risk of stroke or heart attack.
Pheochromocytoma is a rare tumor and doesn’t always cause symptoms. So, it can be difficult to diagnose. Healthcare providers sometimes find pheochromocytomas when you get a test or procedure for another reason.
A provider may suspect a diagnosis of pheochromocytoma after reviewing your medical history and symptoms and doing a physical exam.
Your healthcare provider may use the following tests to diagnose pheochromocytoma:
Advertisement
After your provider diagnoses pheochromocytoma, they’ll likely do additional tests to see if the tumor is local or has spread.
If cancer is found, they’ll stage the cancer. Staging is based on the tumor’s size, if it has spread to nearby lymph nodes or tissues, and if it has spread to distant parts of the body.
If you receive a diagnosis, your provider will recommend genetic testing to determine your risk of having other diseases. If your genetic counselor finds certain gene changes in your testing results, they’ll likely recommend that your family members get testing, too.
Treatment options for pheochromocytoma depend on several factors, including:
The best and first treatment option is surgery, when possible. Surgery successfully removes about 9 in 10 pheochromocytomas.
Surgery involves removing one or both of your adrenal glands. Your surgeon will check the surrounding tissue and lymph nodes to see if the tumor has spread. If it has, your surgeon will remove that, too.
If you have pheochromocytoma that causes symptoms due to excess adrenal hormones, your healthcare provider will likely recommend medication to manage the symptoms before surgery. Medications may include:
Advertisement
Radiation therapy uses beams of energy to destroy cancer cells. The type of radiation therapy your provider may recommend depends on whether the cancer is localized, regional, metastatic or recurrent.
Chemotherapy is a cancer treatment that uses drugs to stop the growth of cancer cells by killing the cells or by preventing them from multiplying.
Ablation therapy uses very high or very low temperatures to destroy tumors. It can help kill cancer cells and abnormal cells.
Embolization therapy blocks the artery leading to your adrenal gland. Blocking the blood flow to your adrenal glands helps kill the cancer cells that are growing there.
Targeted therapy uses medications or other substances to attack specific cancer cells. It helps treat metastatic and recurrent pheochromocytoma. Belzutifan is the first U.S. Food and Drug Administration (FDA)-approved oral targeted therapy for this condition.
Contact your provider if you have new symptoms or your symptoms get worse.
If you’ve recently found out that one of your first-degree relatives (siblings and parents) has a genetic disease, like endocrine neoplasia 2 syndrome or von Hippel-Lindau (VHL) disease, that puts you at a higher risk of developing the tumor. Your provider can talk with you about whether genetic testing makes sense for you.
Advertisement
The outlook for pheochromocytoma is usually good if it’s treated with surgery. After treatment, your provider will check your catecholamine and metanephrine levels. Normal levels are signs that all the pheochromocytoma cells were removed.
Your healthcare provider will recommend follow-up appointments. These visits help make sure your hormone levels stay in a healthy range. Your provider will check for any signs that the tumor has come back.
The five-year survival rate is about 95% when the pheochromocytoma is small and hasn’t spread. If it’s spread or comes back, the five-year survival is 34% to 60%.
Pheochromocytoma and paraganglioma are both rare tumors that come from the same type of cells known as chromaffin cells. Pheochromocytoma is a tumor that forms in the center of your adrenal gland. Paragangliomas form outside of your adrenal gland.
Having a rare tumor can be overwhelming and scary. The good news is that pheochromocytoma is often benign and treatable. Most cases don’t have a known cause. But some are linked to inherited conditions. If you or a first-degree relative has been diagnosed, you should consider genetic testing. This can help identify conditions that may cause other health issues.
If you have any questions about your risk of developing a pheochromocytoma, talk to a healthcare provider. They’re there to help you.


Sign up for our Health Essentials emails for expert guidance on nutrition, fitness, sleep, skin care and more.
Learn more about the Health Library and our editorial process.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
When your hormones are out of balance, so are you. Cleveland Clinic experts can diagnose and treat some adrenal disorders to help you feel like yourself again.
