DiGeorge syndrome (22q11.2 deletion syndrome) is a genetic condition that occurs when your child is missing a piece of chromosome 22. The condition can affect many different areas of your child’s body. They may have unique facial features. DiGeorge syndrome is a lifelong disorder with no cure. But treatment can help manage your child’s symptoms.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
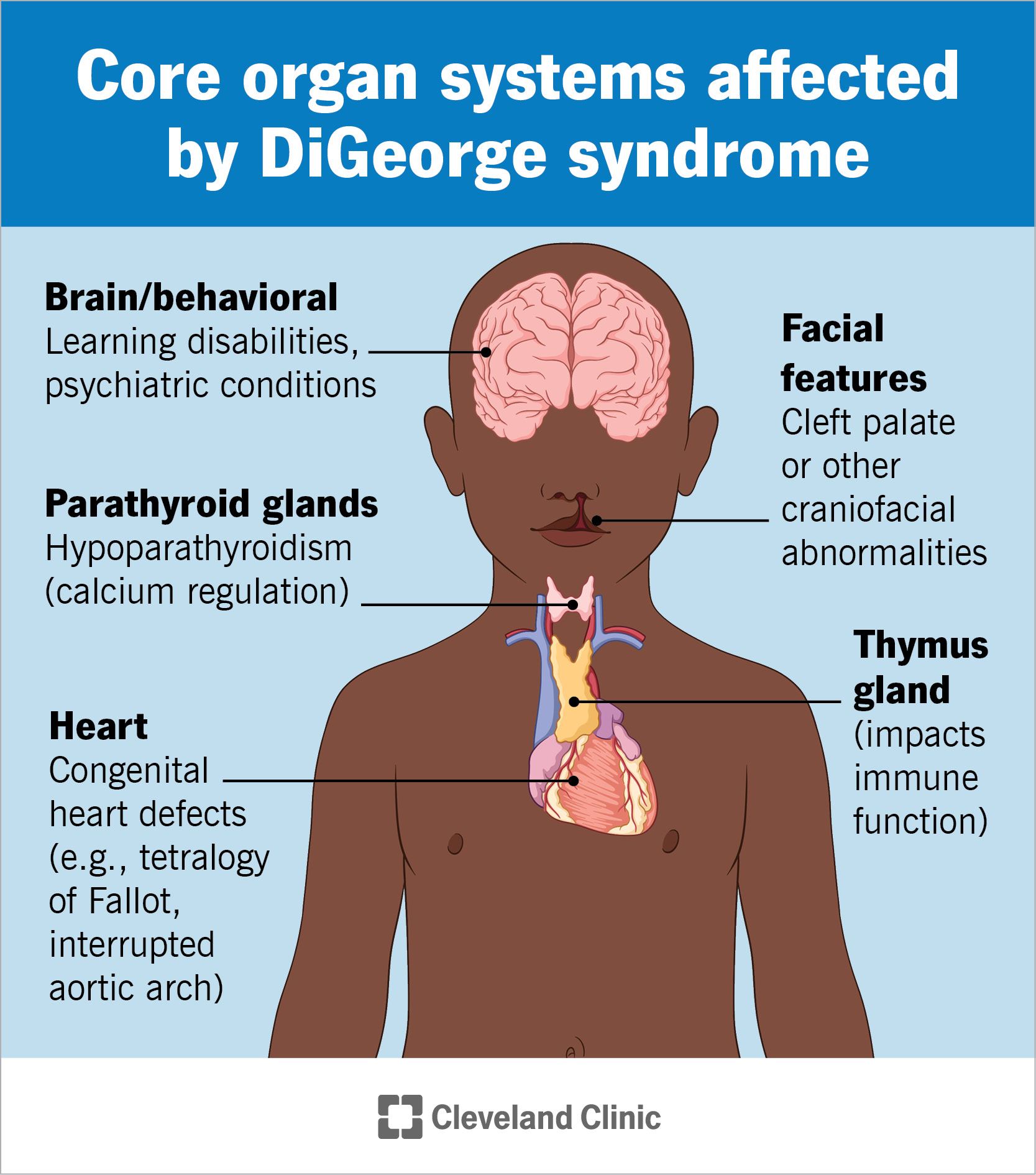
Image content: This image is available to view online.
View image online (https://my.clevelandclinic.org/-/scassets/images/org/health/articles/21182-digeorge-syndrome.jpg)
DiGeorge syndrome is a genetic disorder that can affect many parts of your child’s body. A missing piece of chromosome 22 causes the condition. Common abnormalities associated with DiGeorge syndrome include a cleft palate, kidney anomalies and thymus gland issues. Most children also experience:
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
The complications of DiGeorge syndrome (also known as 22q11.2 deletion syndrome, velocardiofacial syndrome or VCFS) can range from mild to severe. The most severe issues can be life-threatening. There’s no cure for the condition. The diagnosis may feel overwhelming. But treatment can help your child manage the symptoms of this lifelong disorder.
Signs and symptoms of 22q11.2 deletion syndrome vary from child to child. Some children might not experience any symptoms of the condition. Others may have symptoms that affect several parts of their bodies. Your child may experience:
Children with 22q11.2 deletion syndrome may experience life-threatening heart problems, including:
A deletion on chromosome 22 can cause symptoms that affect the function of your child’s immune system. The condition puts them at a higher risk of getting infections. Immune deficiency symptoms may include:
Advertisement
DiGeorge syndrome facial features may include:
22q11.2 deletion syndrome can affect the function and development of your child’s brain. They may experience:
A deletion of part of chromosome 22 can cause other signs and symptoms, including:
A missing part of chromosome 22 causes DiGeorge syndrome. Each chromosome in your child’s body holds thousands of genes. Genes are responsible for providing the instruction manual to help their body grow and function.
The term “22q11.2” gives the specific location on the chromosome where genes are missing. In this condition, they’re missing from segment 11 on the long arm (q) of chromosome 22. When your child is missing genes, their body doesn’t have the instructions it needs to develop as expected. This leads to the symptoms of DiGeorge syndrome, like developmental and cardiac defects.
DiGeorge syndrome can affect anyone. That’s because 9 out of 10 cases occur as a result of a chance deletion on chromosome 22. This happens when the egg and sperm meet in the early stages of fetal development. It happens randomly. You didn’t cause this condition by anything you did before or during pregnancy.
In 1 out of 10 cases, people inherit 22q11.2 deletion syndrome from one biological parent who has the condition. This means they inherit it in an autosomal dominant manner. Only one copy of the chromosome from one parent is necessary for your child to get the condition.
Your pregnancy care provider may be able to make a 22q11 2 deletion syndrome diagnosis before your baby is born. They might find the condition on a prenatal ultrasound or through a prenatal test, like amniocentesis.
If they don’t find the condition before your baby is born, your child’s provider will likely diagnose it soon after birth. They’ll perform a physical exam and look closely at your child’s face, ears and eyes for signs of the condition. They may also request several tests.
Advertisement
Your child’s healthcare provider may use the following tests to diagnose 22q11.2 deletion syndrome:
DiGeorge syndrome treatment depends on the symptoms and conditions that affect your child’s body, often involving multiple medical services. Velocardiofacial syndrome, sometimes referred to as VCFS, often requires a team approach. Treatment may include:
In most cases, your child’s provider will diagnose DiGeorge syndrome when your baby is born or during early childhood checkups. Contact their provider if your child shows signs or symptoms of DiGeorge syndrome.
If your child has trouble breathing, call 911 (or your local emergency service number) or visit the emergency room immediately.
DiGeorge syndrome is a lifelong condition without a cure. Your child’s outlook depends on the severity of their symptoms. Some of their symptoms could be life-threatening, especially if they affect their heart.
Advertisement
With ongoing treatment and support, adults with DiGeorge syndrome can live active and fulfilling lives.
Most people diagnosed with DiGeorge syndrome have a normal life expectancy with mild symptoms. Severe symptoms can lead to a shortened life expectancy without treatment.
It may be overwhelming to learn that your newborn has a genetic condition like DiGeorge syndrome. Remember that their diagnosis isn’t a result of something you did as their parent. Rather, their genetic instruction manual didn’t get completely put together when they were developing.
Talk to your child’s healthcare provider about genetic counseling to learn more about your child’s diagnosis and how you can help them grow.
Advertisement


Sign up for our Health Essentials emails for expert guidance on nutrition, fitness, sleep, skin care and more.
Learn more about the Health Library and our editorial process.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Do certain health conditions seem to run in your family? Are you ready to find out if you’re at risk? Cleveland Clinic’s genetics team can help.
