Polycystic kidney disease (PKD) is a genetic disorder that causes cysts to grow in the kidneys. Health complications include high blood pressure and kidney failure. Most people with PKD will eventually need dialysis or a kidney transplant. PKD affects about 500,000 people in the U.S.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
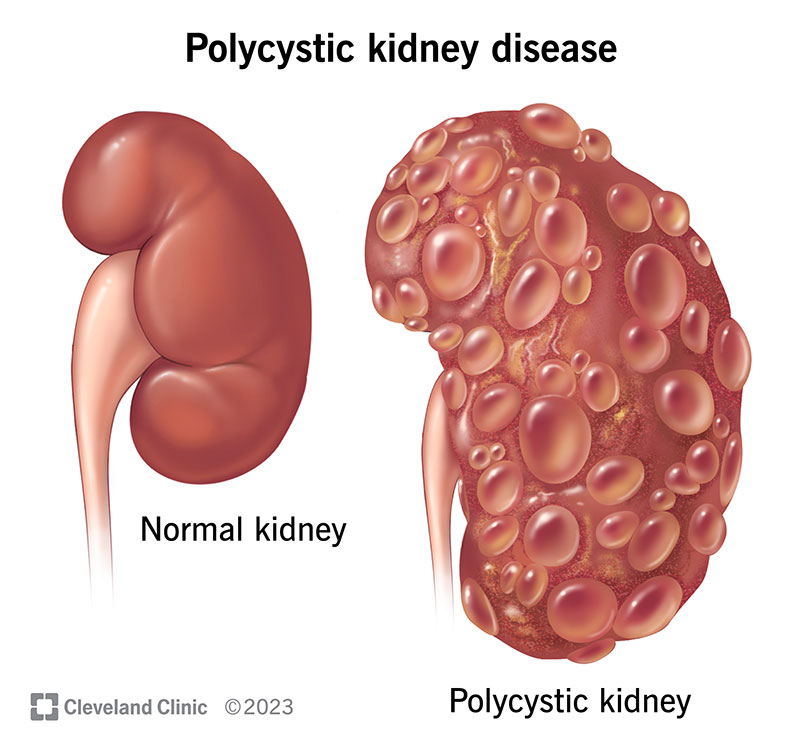
Image content: This image is available to view online.
View image online (https://my.clevelandclinic.org/-/scassets/images/org/health/articles/polycystic-kidney-disease)
Polycystic kidney disease (PKD) causes cysts (fluid-filled growths) to develop in your kidneys. PKD is a genetic disorder, meaning you have to have a mutated (changed) gene to get it.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
PKD isn’t the same as kidney cysts, which are usually harmless. Cysts from PKD can enlarge your kidneys and prevent them from filtering waste out of your blood. In severe cases, the cysts can increase the weight of your kidneys by up to 30 pounds.
PKD causes chronic kidney disease, which can progress to kidney failure. PKD accounts for about 2% of all cases of kidney failure in the United States. Most people with PKD will need dialysis or a kidney transplant.
If you receive a PKD diagnosis, it’s important to work with your healthcare provider on a treatment plan to manage complications of the disease.
There are two types of polycystic kidney disease:
Advertisement
About 500,000 people in the U.S. have PKD.
ADPKD is far more common than ARKPD, affecting about 90% of people with PKD. ARPKD is extremely rare. It occurs in 1 out of 25,000 people.
Symptoms can vary depending on what type of PKD you have.
People with the dominant type of PKD often don’t show signs of the disease until they reach adulthood. You may not have symptoms until the cysts have grown large.
Symptoms of ADPKD include:
People with the recessive type of PKD may show signs of the disease shortly after birth or in childhood. A pregnancy care provider may detect cysts on a fetus’s kidneys during a prenatal ultrasound.
Signs that a newborn may have ARPKD include:
Signs of ARPKD in childhood could include:
Signs that a fetus may have ARPKD include:
No. This disease is often clinically silent, with approximately one-half of cases being diagnosed during the person’s lifetime.
Genetic mutations cause polycystic kidney disease. Genes are the building blocks of your body. You get your genes from your biological parents. They help tell your body how to form and function. Sometimes, a gene mutates and doesn’t copy correctly during fetal development. If you have a gene mutation, you can pass it on to your biological children. This is what happens with most cases of PKD.
In rare cases, genes mutate or change at random without either biological parent carrying the gene.
Since PKD is a genetic condition, you must have a parent that also has the condition.
PKD can cause serious health complications for adults and babies. Some symptoms of PKD are also complications. These include:
Other complications of PKD include:
ARPKD can be fatal in babies who are born with a severe case of the disease. The first month of life is critical for babies born with ARKPD.
A nephrologist (healthcare provider specializing in kidney disorders) usually diagnoses PKD. They may order the following imaging exams to check your kidneys:
Advertisement
A healthcare provider may also recommend genetic testing. A blood or saliva test can check for the mutated genes that cause PKD.
There’s no cure for PKD. The goal of treatment is to slow the progression of the disease and control the symptoms it causes. The most common treatments for PKD include:
Advertisement
People with ADPKD can live long and full lives if they receive the proper treatment and manage the condition. About half of people with ADKPD will need dialysis or a kidney transplant due to kidney failure by age 70.
The outlook for children with ARPKD isn’t as positive. About one-third of all infants born with ARPKD don’t survive. Babies who do survive will likely need medical treatment for the rest of their lives. About half of children who survive infanthood will have kidney failure by age 15 to 20.
Polycystic kidney disease isn’t preventable. But you may be able to slow the disease or prevent kidney failure by practicing a healthy lifestyle. People with PKD who want to have biological children may consider talking to a genetic counselor.
Living a healthy lifestyle can go a long way in managing PKD. Some tips for healthy living include:
Advertisement
Contact a healthcare provider right away if you have PKD and are:
It’s normal to have questions about your diagnosis. Some things you may ask your provider are:
Polycystic kidney disease (PKD) is genetic and causes cysts to form in your kidneys. The cysts may enlarge your kidneys and disrupt how well they work. Most people with PKD are adults. But in rare cases, babies can have a dangerous form of PKD. Treatment reduces symptoms and helps your kidneys work better. It’s important to work with your provider and follow their instructions to manage the condition.


Sign up for our Health Essentials emails for expert guidance on nutrition, fitness, sleep, skin care and more.
Learn more about the Health Library and our editorial process.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Working with healthcare providers who have experience treating polycystic kidney disease is important. Cleveland Clinic’s kidney team is here for you.
