Sickle cell anemia is a form of the inherited blood disorder, sickle cell disease. It’s caused by a genetic variation that turns normal red blood cells into C-shaped or sickle-shaped cells. Symptoms are fatigue from anemia, infections and severe pain. Treatment may ease symptoms and keep the disease from getting worse.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
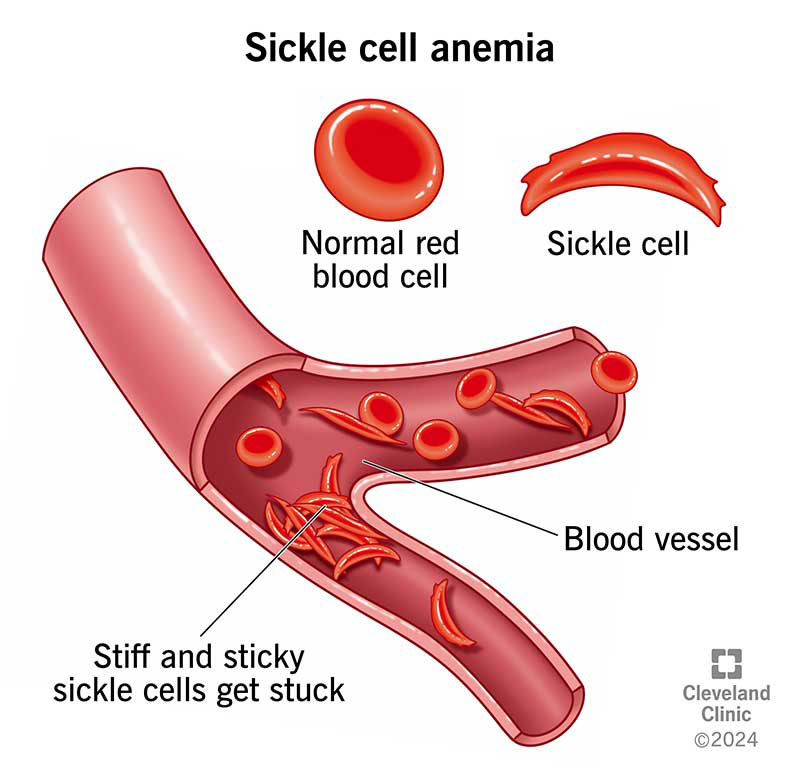
Image content: This image is available to view online.
View image online (https://my.clevelandclinic.org/-/scassets/Images/org/health/articles/sickle-cell-anemia)
Sickle cell anemia is an inherited blood disorder that affects your red blood cells and causes severe anemia. Healthcare providers may refer to these abnormal cells as sickled cells. Sickle cell anemia is the most common and severe type of sickle cell disease.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Normally, your red blood cells are round, flexible disks. They can slide through your blood vessels to carry oxygen to your organs and tissues. Sickled cells are shaped like crescents or sickles and are stiff and sticky. The abnormal shape and form mean sickled cells get stuck in your blood vessels.
Common symptoms include:
The disease’s symptoms typically start when babies are 6 to 9 months old. People’s symptoms may get worse over time as their bodies make more sickled cells.
This disease happens when you inherit genetic variations (changes) that affect the HBB gene. You may be born with sickle cell anemia if both your biological parents carry the changed gene.
The HBB gene gives instructions on how to make beta-globin, which is part of your hemoglobin protein. Hemoglobin helps your red blood cells carry oxygen throughout your body and remove carbon dioxide.
The genetic change means your tissues and organs don’t get enough oxygen. They can’t function without oxygen.
Sickle cell anemia can cause serious and sometimes life-threatening complications. Complications may include:
Advertisement
Healthcare providers may do physical exams that include checking your spleen and liver. They’ll ask about your symptoms, particularly pain in your arms, legs or belly. They may ask about your medical history, including infections. They may order the following tests:
Your treatment depends on your symptoms and your overall health. Common treatments include antibiotics for infections and blood transfusions. Some people may be treated with gene therapy.
There are several medications that can ease sickle cell anemia symptoms and keep the disease from getting worse:
Researchers are testing medications that may reduce how often you have VOC. These newer therapies may also improve red blood cell health and metabolism. They may increase fetal hemoglobin, which would reduce red blood cell sickling.
An allogeneic stem cell transplant is the only cure. You may have this procedure if you have acute chest syndrome, frequent acute pain crises or stroke.
Sickle cell anemia may cause serious medical conditions. Go to the emergency room if you have the following:
Advertisement
The disease also increases your risk of having a stroke. A stroke is a medical emergency. If you have the symptoms below or are with someone who’s having symptoms, call 911 (or your local emergency services number) right away:
This is a chronic disease. This means you or your child likely will always need medical care to manage the disease’s symptoms. The care you’ll need depends on your symptoms. Everyone’s situation is a bit different, so ask your healthcare provider what you can expect. They know your overall health and how sickle cell anemia affects you, so they’re your best source of information.
As you get older, you may develop different and more serious medical problems that happen when organ tissues don’t receive enough oxygen. People with sickle cell anemia are at an increased risk of stroke. The disease also increases the risk of damage to their lungs, kidneys, spleen, eyes and liver — among other organs.
Advertisement
Thanks to early diagnosis and treatment to ease complications, people with this disease may live into their 50s. Some people may live much longer. If you have questions about how long you may live, ask your healthcare provider. They’ll explain how the disease may affect how long you’ll live.
Sickle cell anemia is a challenging condition that can affect you in many ways. Some suggested ways to feel better include:
Advertisement
There was a time when few babies born with sickle cell anemia lived long enough to celebrate their 5th birthdays. Now, people are living much longer with this disease. But this condition is still a serious illness that can cause life-threatening complications.
If you or your child has sickle cell anemia, you may be grateful for the past progress, but hopeful there will be new treatments. Researchers are testing new ways to treat this condition. Ask your provider if a clinical trial is right for you.


Sign up for our Health Essentials emails for expert guidance on nutrition, fitness, sleep, skin care and more.
Learn more about the Health Library and our editorial process.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic cares for people of all ages with sickle cell disease. We walk you through your diagnosis and treatment and offer education and support services.
