Chordoma is a rare bone tumor that forms in your spine or skull base. All subtypes of chordoma are considered cancerous (malignant). Surgical removal of the tumor is the first-line treatment, but it can be difficult to completely remove chordomas due to their location near your spinal cord or brainstem.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
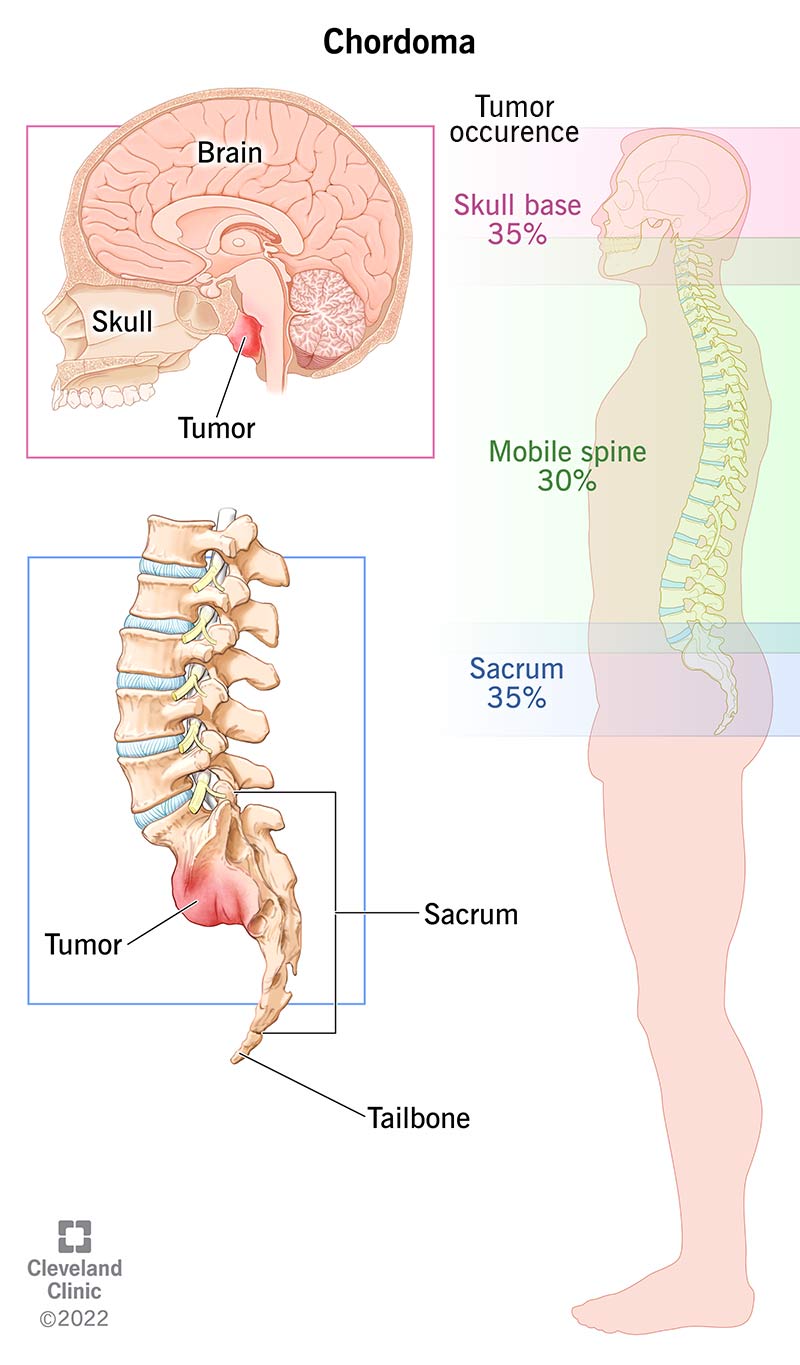
Image content: This image is available to view online.
View image online (https://my.clevelandclinic.org/-/scassets/images/org/health/articles/17916-chordoma)
Chordoma is a rare malignant (cancerous) bone tumor that forms in your spine or the base of your skull. It’s a type of sarcoma.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Chordomas can occur at any point along your spine:
Chordomas typically grow slowly, but they can be difficult to treat due to how they invade nearby nervous system tissue.
They also tend to recur (come back) after treatment — usually in the same place. Chordomas spread to other parts of your body (metastasize) in 30% to 40% of cases.
If an advanced chordoma does metastasize, it most commonly spreads to your:
The World Health Organization (WHO) recognizes three distinct types of chordoma based on what the cells look like under a microscope (their histology):
Advertisement
Chordomas can develop in anyone at any age, but they’re most likely to occur in adults aged 50 to 80. About 5% of cases affect children.
Men are approximately 1.5 times as likely to have a chordoma as women.
Chordomas are rare. They affect about 1 person for every 1 million people per year. That means that about 300 people receive a chordoma diagnosis each year in the United States.
About 1% to 4% of all primary bone tumors are chordomas.
As a chordoma grows, it puts pressure on nearby areas of your spinal cord or brain. This pressure causes the symptoms of chordoma. Symptoms can also vary based on where the tumor is along your spine.
General chordoma symptoms include pain, weakness and/or numbness in your back, arms or legs.
Symptoms of a chordoma at the base of your skull may include:
Symptoms of a chordoma in your tailbone may include:
Researchers don’t know exactly why chordomas form. But they think changes (mutations) in the TBXT gene are likely involved.
A few families have had multiple members who’ve developed a chordoma. Studies revealed that these people inherited a duplication of the TBXT gene. Researchers have also identified changes in the TBXT gene in people with chordoma who have no family history of the condition.
A chordoma develops from cells of the notochord. This is a structure that’s present in a developing embryo and is important for the future development of its spine. The notochord usually disappears by the time the fetus is eight weeks old. But in a small percentage of people, a few notochord cells can remain embedded in the bones of the spine or the base of the skull.
A change in the TBXT gene may trigger the growth of these cells, leading to a chordoma.
People with a genetic condition called tuberous sclerosis are at higher risk of developing chordoma. This condition causes a variety of medical issues, including epilepsy, developmental delay and tumors throughout your body. Tuberous sclerosis is caused by mutations in two genes: TSC1 and TSC2.
Your healthcare provider will ask about your symptoms and medical history. They’ll likely perform a physical exam and a neurological exam.
If they suspect a tumor, they’ll order an imaging test, such as an X-ray, computed tomography (CT) scan or MRI scan.
Your provider will likely refer you to a bone cancer specialist for a second opinion and confirmation of the diagnosis. You may need additional imaging tests to better determine the location of the chordoma and see if it’s spread to other areas of your body.
Advertisement
The only way healthcare providers can definitively diagnose chordoma is with a biopsy — usually a needle biopsy. This involves taking a small sample of the tumor so a specialist can examine it under a microscope.
The go-to treatment option for chordoma is surgery. Total surgical removal of the tumor (en bloc resection) has the best chance of prolonging survival. However, this is often difficult due to the location of the tumors. Specifically, this isn’t possible for chordomas in the base of your skull.
A chordoma in your spine can invade your spinal cord and nearby important nerves and arteries, which could cause lasting issues or death if they’re damaged during surgery. A chordoma at the base of your skull is often difficult to completely remove because it’s close to essential structures such as your brainstem, cranial nerves and spinal cord. Neurosurgeons aim to remove as much of the chordoma as they safely can.
Chordomas are generally resistant to radiation therapy and chemotherapy as primary treatments. But your healthcare team might recommend radiation therapy after surgery to lower the chance that the tumor will grow back.
Researchers are currently studying experimental therapies for chordomas, such as targeted therapy and immunotherapy. There may be clinical trials available that you can participate in.
Advertisement
The prognosis (outlook) of chordoma varies depending on a few factors:
Your healthcare team will be able to give you more accurate information about what you can expect. Don’t be afraid to ask them questions.
Yes, chordoma can cause death — typically due to tissue destruction in your spinal cord, brain or brainstem after it has recurred (come back).
A study of 357 people with chordoma revealed that the survival rates were:
It’s important to remember that these are just averages. Your healthcare team can provide more detailed information about survival rates based on your unique situation.
There’s nothing you can do to prevent developing chordoma. Most cases happen randomly.
Advertisement
If you have a family history of chordoma or tuberous sclerosis, be sure to see your healthcare team regularly so they can monitor you for signs of chordoma. The earlier they can catch it, the better.
Chordomas often come back (recur), even many years after treatment. Because of this, long-term follow-up with your healthcare team is important.
If you have any new or worsening symptoms, talk to your healthcare provider.
Chordoma is a type of bone cancer. More specifically, it’s a type of sarcoma, which is a broad group of cancers that begin in your bones and your soft (connective) tissues.
No. All subtypes of chordoma are considered malignant (cancerous).
Learning you have a rare type of bone cancer can be scary and stressful. Chordomas can be difficult to treat because of their location. But know that your healthcare team will develop an individualized and thorough treatment plan to help treat the chordoma and improve your quality of life.


Sign up for our Health Essentials emails for expert guidance on nutrition, fitness, sleep, skin care and more.
Learn more about the Health Library and our editorial process.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
When you learn you have a skull base tumor, you’ll want the best care from the best providers. Cleveland Clinic is here to help.
