Rett syndrome is a rare neurological condition that affects your child’s movement and growth. A sporadic (random) gene variant causes it. Almost all children diagnosed with Rett syndrome need caregiver support throughout their lives to manage symptoms.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Rett syndrome is a rare neurodevelopmental condition that mainly affects girls. It leads to symptoms related to movement, communication and thinking abilities. A genetic variant that affects brain development causes the syndrome.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
The first signs of the condition appear around 6 months of age. As a parent or caregiver, you might suspect something’s wrong when your child stops meeting developmental milestones. The symptoms typically show up in stages as your child gets older.
There’s no cure for Rett syndrome. Symptoms that affect your child’s development often worsen early on, then stabilize over time. They don’t go away. Certain medications and therapies can help manage the symptoms.
Rett syndrome progresses in stages as your child gets older. They might experience different symptoms at each stage:
Advertisement
Not all children go through every stage. For example, some people with Rett syndrome are never able to walk.
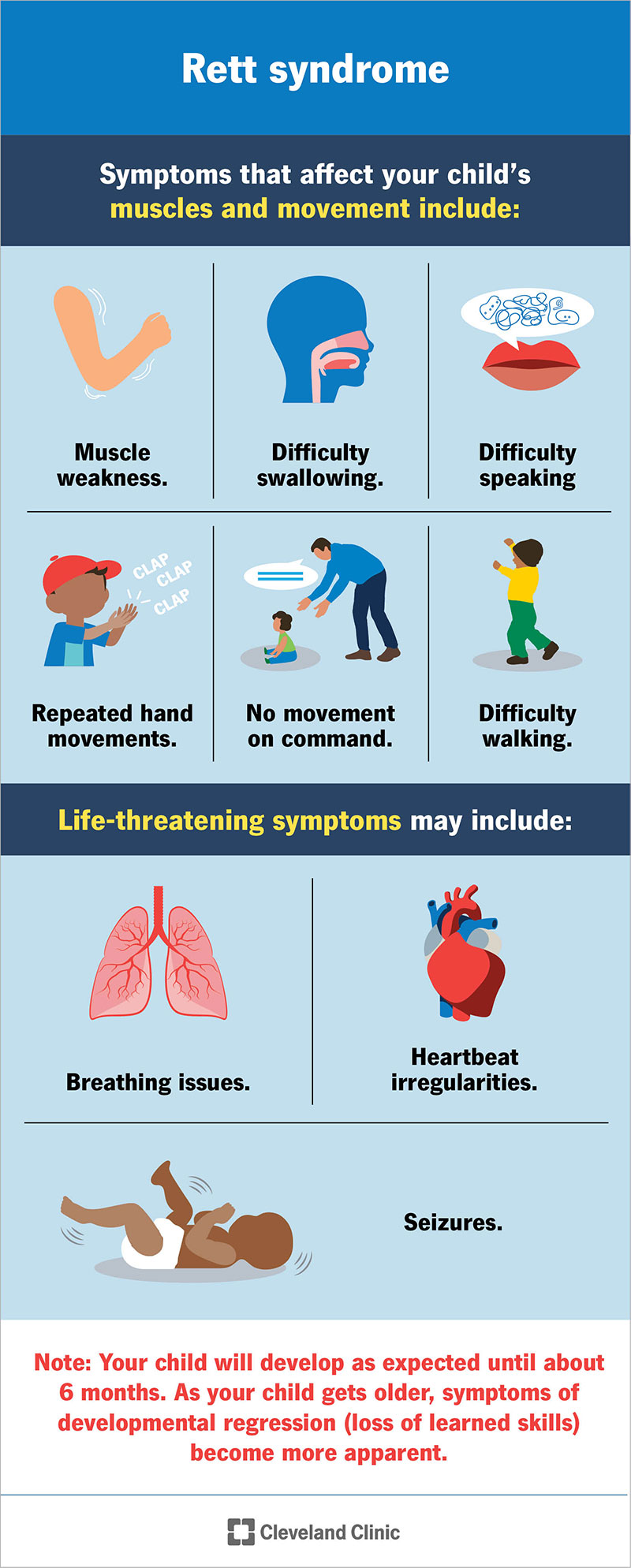
Image content: This image is available to view online.
View image online (https://my.clevelandclinic.org/-/scassets/images/org/health/articles/rett-syndrome)
With Rett syndrome, your child develops as expected until about 6 months. Then, you may notice developmental delays. These happen when your child doesn’t reach expected milestones for their age. Examples of milestones include waving, walking and saying their first words.
As your child gets older, the loss of learned skills (developmental regression) becomes more visible.
Symptoms vary from child to child but may include:
A genetic variant of the MECP2 gene causes most cases of Rett syndrome. This gene provides instructions to make the MECP2 protein. This protein helps nerve cells connect and communicate. It helps your child’s brain function as expected.
Not all cases of Rett syndrome involve the MECP2 gene. Issues with other genes, like CDKL5 and FOXG1, can cause atypical forms. Some unidentified genes can also cause symptoms.
The genetic change occurs spontaneously (randomly). It’s not usually inherited. There’s nothing you can do to prevent this syndrome.
Rett syndrome almost always affects females. This is because the genetic change that causes it happens on the X chromosome. You have two X chromosomes if you’re female.
As males have one X chromosome and one Y chromosome, this condition rarely affects them. A variant on a child’s only X chromosome can lead to miscarriage or death during early infancy due to severe symptoms.
Healthcare providers identify this gene change in males as MECP2-related severe neonatal encephalopathy. This condition can cause similar symptoms, like intellectual disability, seizures and difficulty with movement.
Your child may be at risk of the following complications, some of which are life-threatening:
Healthcare providers use a physical exam and testing to diagnose Rett syndrome. A diagnosis usually happens after 6 months of age and before 18 months.
Advertisement
Your child’s provider will use tests to rule out more common conditions with similar symptoms. A genetic blood test can confirm a Rett syndrome diagnosis in most cases. This genetic screening doesn’t require any special preparation or a hospital stay.
As Rett syndrome is so rare, a diagnosis usually doesn’t happen quickly. This can be frustrating as you wait for answers. But a clear diagnosis helps your child’s care team treat their symptoms.
There isn’t a cure for Rett syndrome. But your child’s healthcare providers can help manage their symptoms throughout their life.
Treatment varies based on the specific symptoms. For example, medications can treat seizures and movement challenges. If your child has difficulty with motor skills and language, their provider may recommend:
Your child may also benefit from:
A medication called trofinetide is the first U.S. Food and Drug Administration (FDA)-approved treatment specifically for Rett Syndrome. The medication is for children 2 years of age and older. It’s not a cure. It’s a disease-modifying treatment. You’ll need to discuss and work closely with your medical team to explore this option.
Advertisement
Many people with Rett syndrome live a high quality of life well into their 40s and beyond. If symptoms are mild, your child may have a typical life expectancy. Health complications may shorten life expectancy.
Your child’s provider is the best person to ask about their life expectancy. They understand and can explain the outlook of your child’s unique situation.
Rett syndrome is a lifelong condition. Symptoms affect each person differently. Your child may be able to manage their movements, walk and communicate on their own. But they’ll still need around-the-clock care throughout their life.
Your child will also need regular visits with their care team to check on symptoms and prevent complications. Some cases of Rett syndrome can cause early death if complications arise.
It can be challenging to learn that your child has a rare neurological condition. Your child will need more care as they grow. But you don’t have to do it all on your own. Your child’s care team will be with you every step of the way.
Researchers, through clinical trials, are finding new treatment options to help children diagnosed with Rett syndrome. If you have any questions about your child’s outlook or treatment, let their providers know.
Advertisement


Sign up for our Health Essentials emails for expert guidance on nutrition, fitness, sleep, skin care and more.
Learn more about the Health Library and our editorial process.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
When your child has a neurological condition, you want them to have the best care. At Cleveland Clinic Children’s, we offer compassionate, personalized treatment.
