Creutzfeldt-Jakob disease (CJD) is a rare and severe degenerative brain condition. It happens when abnormal proteins, known as prions, damage your brain. This condition usually worsens very quickly. Most people don’t survive more than a year after diagnosis. Supportive care is available.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Creutzfeldt-Jakob (pronounced “croy-tz-felt” “yah-cob”) disease (CJD) is a rare, degenerative brain disorder caused by abnormal proteins called prions. These proteins build up in brain cells and destroy them. As a result, CJD causes dementia symptoms, such as memory loss and confusion, and may also cause behavioral and movement changes.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
CJD is always fatal. There’s no cure or way to slow its progression. Care and support options are available to help you stay comfortable.
It’s a very rare disease, with 1 to 2 cases happening in every 1 million people worldwide. About 350 people receive a diagnosis of CJD in the U.S. each year.
A well-known but also very rare type is variant CJD (vCJD). It’s caused by eating beef from cattle infected with bovine spongiform encephalopathy (BSE), commonly known as “mad cow disease.”
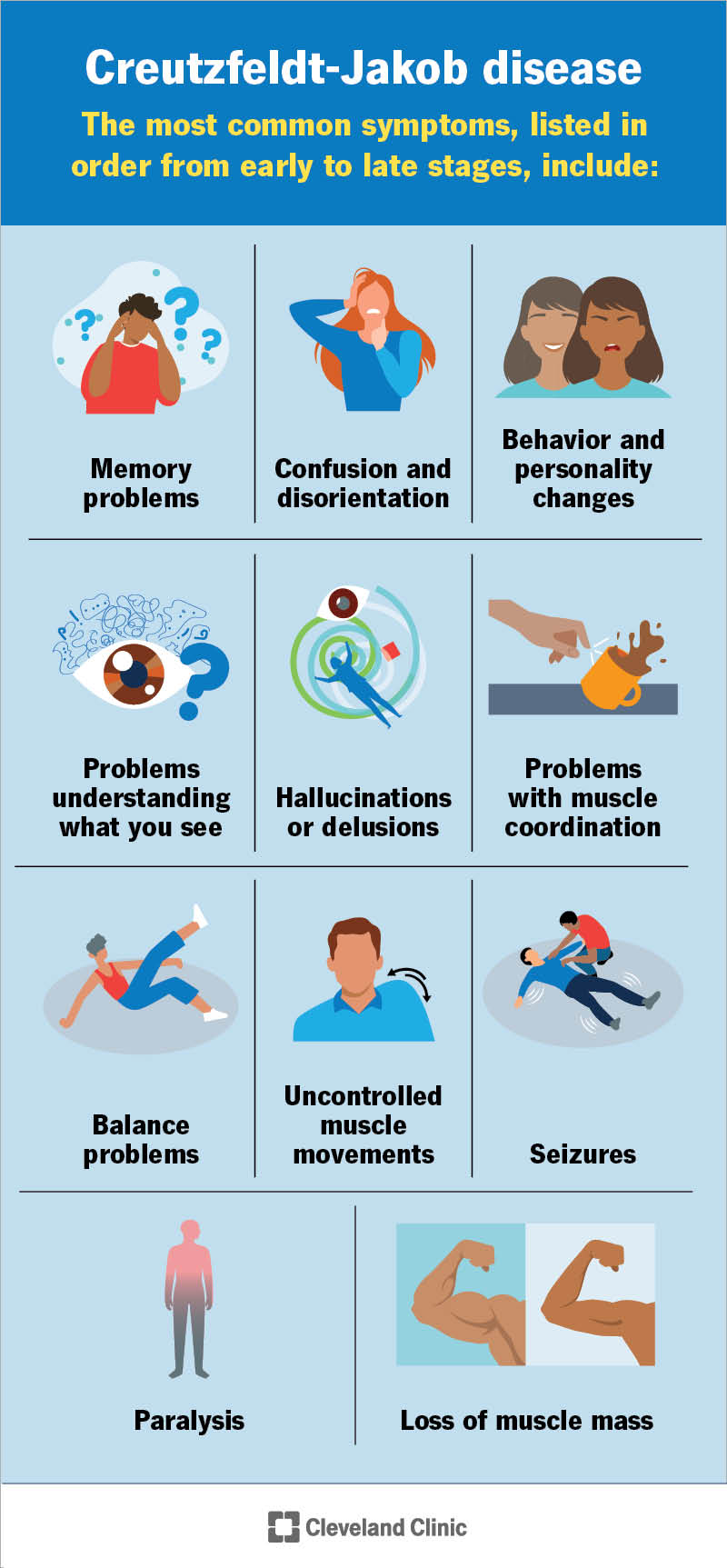
Image content: This image is available to view online.
View image online (https://my.clevelandclinic.org/-/scassets/images/org/health/articles/creutzfeldt-jakob-disease)
The most common symptoms of CJD, listed in order from early to late stages, include:
Abnormal proteins called prions cause Creutzfeldt-Jakob disease. Proteins are molecules — tiny particles that need to hold a specific shape to function. They fold like origami. Sometimes, the folding process goes wrong. When a protein misfolds, your brain cells can’t use it.
Advertisement
As your cells can’t get rid of misfolded proteins, they build up. In the case of prions, the buildup damages and eventually destroys brain cells. In addition, prions are infectious. They trigger normal proteins to misfold, creating even more prions. As they multiply and spread, the disease progresses rapidly.
Types of CJD include:
Creutzfeldt-Jakob disease can affect anyone. You may have prions for years before symptoms appear. But when they do start, symptoms progress very quickly as the disease damages and destroys more of your brain.
It typically affects people between the ages 50 and 80. However, the genetic types usually happen between age 30 and 50.
CJD isn’t easily contagious from person-to-person contact. The only way to spread it from person to person is through organ or tissue transplants or certain types of hormones taken from a donor who had CJD.
Variant CJD may be passed in blood transfusions, but this is extremely rare. Variant CJD is also extremely rare on its own.
A healthcare provider will diagnose CJD by looking for signs and symptoms during a physical and neurological exam. They may ask you to do certain tasks, which can help them identify problems with how your brain functions.
Experts classify CJD as a type of transmissible spongiform encephalopathy. This refers to how your brain with prion damage looks under a microscope (full of holes, like a sponge). Your provider will review tests, like an MRI or spinal tap (lumbar puncture), to confirm a CJD diagnosis.
Your provider may use other tests to evaluate for CJD and conditions with similar symptoms:
There’s no way to cure, treat or slow the progress of CJD. Your provider may offer options for symptom relief (palliative care). Some examples may include medications to help with seizures, behavioral changes or uncontrollable muscle jerks. However, these treatments offer limited benefits.
Advertisement
This condition worsens quickly. Supportive care is available for you and your loved ones. During the late stages, hospice services can be very helpful.
Your care team might recommend participating in a clinical trial if you qualify. Clinical trials help providers study new treatment options.
CJD has a very poor outlook because the condition isn’t curable or treatable. After symptoms start, you’ll soon lose the ability to care for yourself, move and communicate.
As changes can happen fast, it’s important to work with your care team to make sure your wishes and needs are met. Everyone, whether you currently have an illness or not, should complete an advance directive document to tell your providers what type of care you’d like to receive when you’re unable to tell them yourself. Completing an advance directive is even more important in the early stages of a rapidly progressive disease like CJD.
Because this condition is so severe, you and your loved ones should consider speaking with a mental health professional to help you cope with the effects and changes that are to come.
Most cases of CJD are fatal within months to a year after diagnosis. The exception is genetic CJD, which can have a survival time of one to 10 years after symptoms start.
Advertisement
Your provider can give you the most up-to-date information on your case specifically. Remember that individuals are not statistics.
Most cases of CJD can’t be prevented. The exception is variant CJD. This comes from eating beef from cows with bovine spongiform encephalopathy (BSE).
Animal inspections help keep BSE-infected cattle out of the food supply. But uninspected or improperly processed meat can still pose a risk. To stay safe, avoid eating uninspected or unregulated sources of meat, especially brain tissue or bone marrow.
A Creutzfeldt-Jakob disease (CJD) diagnosis is life-changing. It’s like you’re losing your connection to yourself and the world around you in the blink of an eye. That kind of change is difficult on both you and the people closest to you. If you’re unsure what to expect or how to move forward, your care team is just a question away. They can help you plan ahead and feel more prepared for what’s to come.
Advertisement


Sign up for our Health Essentials emails for expert guidance on nutrition, fitness, sleep, skin care and more.
Learn more about the Health Library and our editorial process.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Memory loss, dementia and other cognitive disorders need personalized treatment for the best results. Cleveland Clinic’s brain health experts are here to help.
