Wilson disease is a rare genetic condition where copper builds up in your body. The condition affects your liver, brain, eyes and other organs. Too much copper in your body can cause life-threatening organ damage without treatment.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Wilson disease is a rare genetic condition that occurs when your body accumulates too much copper, especially in the liver and brain. Your body needs a small amount of copper from food to stay healthy, but without treatment, Wilson disease can lead to high copper levels that cause life-threatening organ damage.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Wilson disease is passed on from parents to their children. It requires a copy of the abnormal gene from each parent. It’s very difficult to know who will develop it because parents often have no symptoms to let them know that they carry the abnormal gene. If you have other close relatives with the disease, you’re more likely to be at risk.
Wilson disease affects an estimated 1 out of every 30,000 people. It’s more common among people with a family history of the condition. There are more people who have only one copy of the abnormal gene (carriers). Carriers won’t usually have symptoms, even though they can give the disease to their children. Because there are no symptoms in carriers, it’s hard to know exactly how many people in the general population have an abnormal copy of the gene.
Wilson disease, without early diagnosis and treatment, can lead to life-threatening complications. Toxic levels of copper build up in your body, especially in the liver and brain, and put you at risk of organ damage. Increased copper levels will affect how you feel, often making you very tired or weak and uncomfortable or in pain. If you have weakness, tiredness or pain that doesn’t go away, reach out to your healthcare provider for advice.
Advertisement
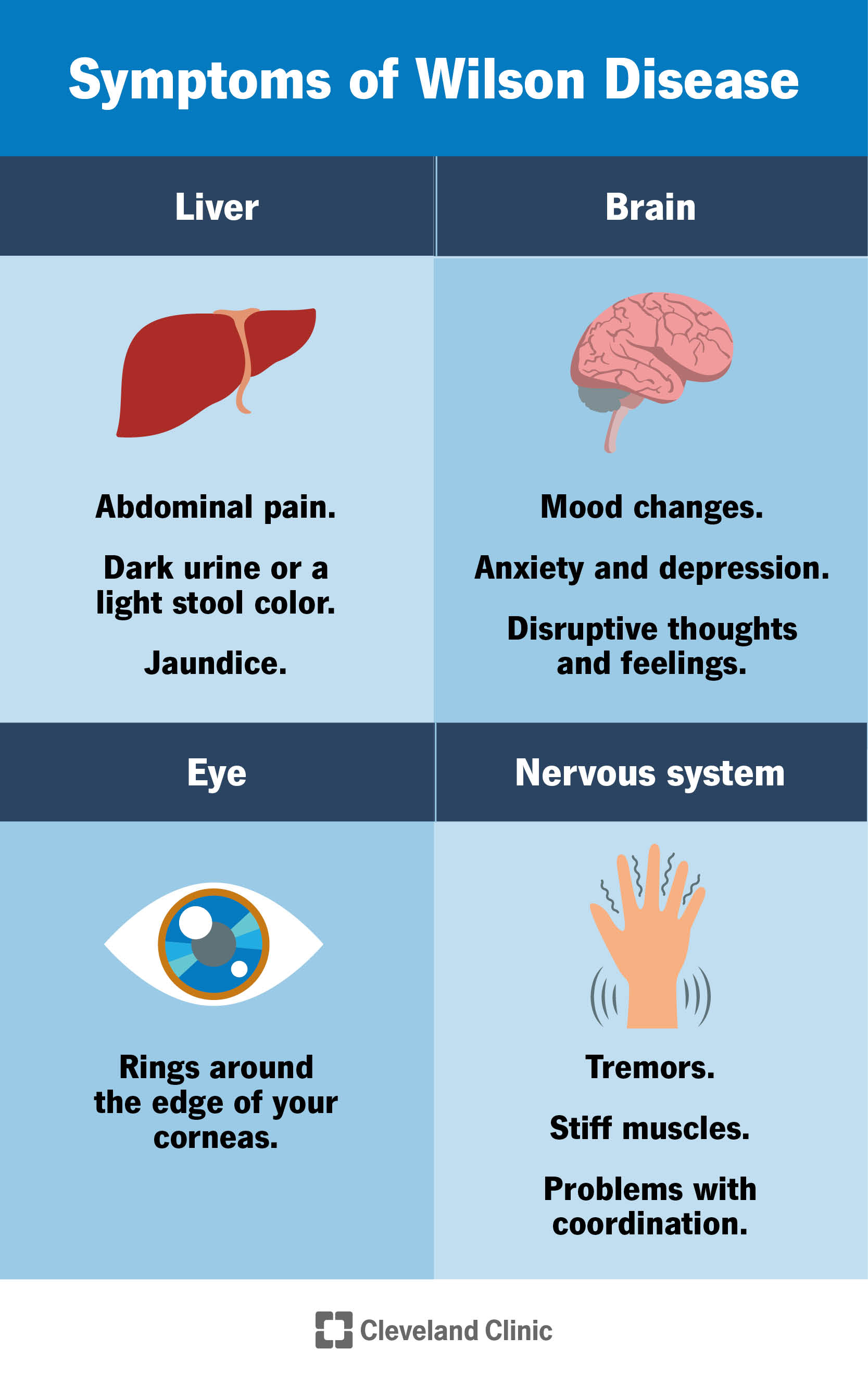
Image content: This image is available to view online.
View image online (https://my.clevelandclinic.org/-/scassets/images/org/health/articles/5957-wilson-disease)
Symptoms of Wilson disease vary a lot from person to person.
Wilson disease is present at birth (congenital), but the symptoms don’t appear until copper builds up in your liver, brain, eyes or other organs. People who have Wilson disease typically develop symptoms between ages 5 and 40. However, some people develop symptoms at younger or older ages.
Some people are diagnosed with other liver or mental health disorders when they actually have Wilson disease. This is because symptoms can be nonspecific and similar to other conditions until copper levels are measured.
People with Wilson disease often develop symptoms of hepatitis (inflammation of the liver) and can have an abrupt decrease in liver function (acute liver failure). These symptoms may include:
Some people with Wilson disease have symptoms only if they develop chronic liver disease and complications from cirrhosis. These symptoms may include:
People with Wilson disease may develop central nervous system symptoms that affect their mental health as copper builds up in their body. These symptoms are more common in adults but do also occur in children.
Nervous system symptoms may include:
Symptoms of Wilson disease that affect a person’s mental health include:
Many people with Wilson disease have green, gold or brown rings around the edge of the corneas in their eyes (Kayser-Fleischer rings). A buildup of copper in the eyes causes the Kayser-Fleischer rings. Your healthcare provider can see these rings during a special eye exam called a slit-lamp exam.
Most people diagnosed with Wilson disease who have symptoms that affect their nervous system also have Kayser-Fleischer rings. About half of the people diagnosed with Wilson disease who only have symptoms that affect their liver also have Kayser-Fleischer rings.
Wilson disease can affect other parts of your body and cause symptoms including:
Advertisement
A mutation of the ATP7B gene causes Wilson disease. This gene is responsible for removing extra copper from your body.
Normally, the liver releases extra copper into a fluid that’s then stored in your gallbladder to help digestion (bile). Bile carries copper, along with other toxins and waste products, out of the body through your digestive tract. If you have Wilson disease, your liver releases less copper into your bile, and the extra copper stays in your body.
You can inherit the mutation of the ATP7B gene that causes Wilson disease. This means that the mutated gene passes from parent to child. To get Wilson disease, a person must inherit two abnormal genes, one from each parent (autosomal recessive).
People who have one ATP7B gene without a mutation and one ATP7B gene with a mutation don’t have Wilson disease, but they’re carriers of the disease. This means that they can pass a normal gene, carrier state or disease state to their children, depending on the genetics of their partner.
To diagnose Wilson disease, your healthcare provider will ask about your family history and personal medical history to identify whether symptoms could be the result of this condition.
Advertisement
Providers will examine you for other signs that tell them you have problems with your liver, brain or eyes. During an eye exam, your provider will perform a slit-lamp exam, which is a special light that looks for Kayser-Fleischer rings in your eyes.
If they suspect Wilson disease, your healthcare provider will want to do blood and urine tests.
The diagnosis of Wilson disease is made using blood tests, urine tests, genetic testing or liver biopsy.
Blood tests can look at many substances in your blood including:
Your healthcare provider may order a blood test to check for the genetic mutation that causes Wilson disease if other medical tests don’t confirm or rule out a diagnosis of the condition.
For 24 hours, you will collect your urine at home in a special container that’s copper-free, provided by your healthcare provider. A lab will check the amount of copper in your urine. Copper levels in the urine are higher than normal in people who have Wilson disease.
Advertisement
If the results of blood and urine tests don’t confirm or rule out a diagnosis of Wilson disease, your provider may order a liver biopsy. During a liver biopsy, your healthcare provider will take a small sample of tissue from your liver. A pathologist will examine the tissue under a microscope to look for features of specific liver diseases, such as Wilson disease, and check for liver damage and cirrhosis. A piece of liver tissue will be sent to a lab, which will check the amount of copper in the tissue.
If you have nervous system symptoms, your healthcare provider may use imaging tests to check for signs of Wilson disease or other conditions in the brain. Tests could include:
Treatment for Wilson disease focuses on lowering toxic levels of copper in your body and preventing organ damage and the symptoms you get when your organs aren’t functioning normally. Treatment includes:
People who have Wilson disease need lifelong treatment. Stopping treatment may cause acute liver failure. Your healthcare provider will regularly perform blood and urine tests to check how the treatment is working.
Your healthcare provider may offer several different medications to reduce the amount of copper in your body.
Chelating agents remove copper from your body. Types of chelating agents include:
When treatment begins, doctors gradually increase the dose of chelating agents. People take higher doses of chelating agents until the extra copper in their body goes away. When Wilson disease symptoms improve and tests show that copper is at safe levels, your provider may prescribe lower doses of chelating agents as maintenance treatment. Lifelong maintenance treatment prevents copper from building up again.
These medications can have side effects and you should ask your provider if you need to take dietary supplements like vitamins or take any precautions before having surgery.
Zinc prevents the intestines from absorbing copper. Your healthcare provider may prescribe zinc as a maintenance treatment after chelating agents remove extra copper from your body. Your provider may also prescribe zinc if you have Wilson disease but don’t have symptoms.
If you’re pregnant, you should ask your provider how to continue treatment for Wilson disease throughout your pregnancy. Your provider might prescribe a lower dose of chelating agents because the fetus needs a small amount of copper. Having an obstetrician who is familiar with Wilson disease can be very helpful.
Also ask your provider if it’s safe to breastfeed while you’re being treated for Wilson disease.
Your healthcare provider may recommend changing your diet to avoid certain foods that are high in copper if you have Wilson disease. You should specifically avoid:
Other foods that are high in copper include:
After treatments lower your copper levels and you begin maintenance treatment, talk with your healthcare provider about whether you can safely eat moderate amounts of some of these foods.
If your tap water comes from a well or runs through copper pipes, have the copper levels in your water checked. You may need to use a water filter to remove copper from your tap water.
If you plan on taking dietary supplements like vitamins, talk with your healthcare provider before taking them. Some dietary supplements contain copper.
Wilson disease may lead to complications, but early diagnosis and treatment can lower your chances of developing side effects. The medications used to treat Wilson disease also may have side effects, so ask your provider what to watch for.
Wilson disease can cause acute liver failure, a condition where your liver quickly stops functioning without warning. About 5% of people with Wilson disease have acute liver failure when they receive their diagnosis. Acute liver failure could require a liver transplant.
Acute kidney failure and a type of anemia called hemolytic anemia often occur in people who have acute liver failure due to Wilson disease.
Cirrhosis is a condition where scar tissue replaces healthy liver tissue and prevents your liver from working normally. Scar tissue also partially blocks the flow of blood through your liver. As cirrhosis gets worse, the liver begins to fail.
Among people diagnosed with Wilson disease, 35% to 45% already have cirrhosis at the time of diagnosis.
Cirrhosis increases your chance of getting liver cancer. However, healthcare providers found that liver cancer is less common in people who have cirrhosis due to Wilson disease than in people who have cirrhosis due to other causes.
Cirrhosis may eventually lead to liver failure. With liver failure, your liver is badly damaged and stops working. Liver failure is also called end-stage liver disease. This condition may require a liver transplant.
If Wilson disease leads to cirrhosis, your healthcare provider may be able to treat your complications with medicine or surgery.
If Wilson disease causes acute liver failure or chronic liver failure due to cirrhosis, you may need a liver transplant. Some people who receive a liver transplant will completely recover from Wilson disease, but your healthcare provider will carefully monitor you to make sure the transplant was successful.
Treatment for Wilson disease is lifelong. The timeline for when you’ll feel better depends on the severity of your symptoms, but your symptoms could reduce significantly after four to six months of treatment followed by regular maintenance treatment. Wilson disease requires regular testing by your healthcare provider to understand if the treatment is successful and they will adjust your medicine dosage to meet your body’s needs.
Depending on your diagnosis, you may or may not experience symptoms associated with Wilson disease. If you do experience symptoms, they could be life-threatening if you don’t receive treatment. It’s important to stick to your healthcare provider’s treatment plan to remove toxic copper from your body and prevent damage to your organs.
Treatment for Wilson disease is lifelong and there’s no cure. The life expectancy is short for someone with Wilson disease who doesn’t receive treatment. For people diagnosed with Wilson disease who successfully receive a liver transplant, the prognosis is good but you will also need to be monitored lifelong after the transplant.
You can’t prevent Wilson disease since it’s the result of an inherited genetic mutation. If you have a family history of Wilson disease, talk with your healthcare provider about genetic testing to understand your risk of developing Wilson disease or having a child with this genetic condition.
If you have Wilson disease, you can take care of yourself by taking medicine as directed by your provider and by removing foods that are high in copper from your diet. With treatment, you’ll be able to reduce your symptoms and prevent life-threatening complications.
You should visit your healthcare provider if your symptoms of Wilson disease get worse, especially if they previously improved. This could mean that your medicine dosage needs to change.
Visit the emergency room immediately or call 911 if your symptoms of Wilson disease prevent you from going about your normal routine, especially if you can’t eat, have persistent vomiting, you experience psychosis, your skin turns yellow (jaundice) or you have severe abdominal pain.
Early diagnosis and treatment of Wilson disease lead to the best outcome for people with this lifelong genetic condition. You may need to make lifestyle changes to remove copper from your diet. Stay up to date on visits to your healthcare provider to make sure your treatment is successful at lowering the amount of copper in your body. The right treatment will help you feel better and avoid life-threatening complications.


Sign up for our Health Essentials emails for expert guidance on nutrition, fitness, sleep, skin care and more.
Learn more about the Health Library and our editorial process.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic’s primary care providers offer lifelong medical care. From sinus infections and high blood pressure to preventive screening, we’re here for you.
