Hurler syndrome is the most severe form of mucopolysaccharidosis type I, a hereditary lysosomal storage condition. Cells can't break down sugar molecules, which affects how they function. Symptoms are life-threatening, target your bones and organs, and could cause issues with cognitive development. Treatment increases life expectancy.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
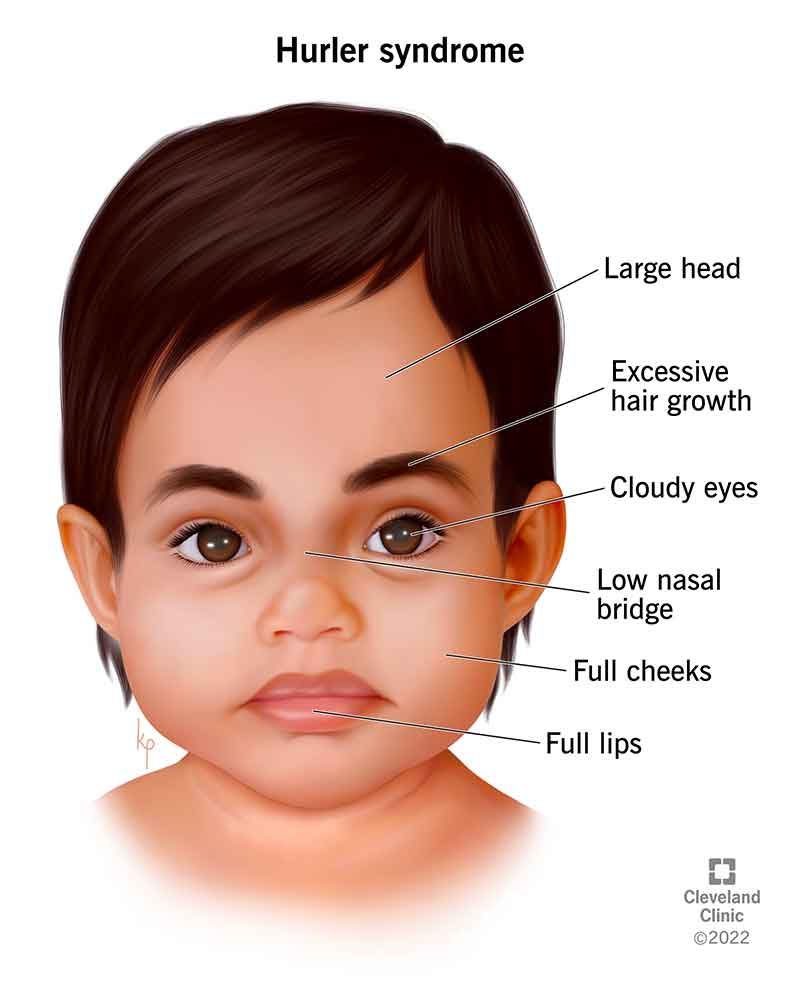
Image content: This image is available to view online.
View image online (https://my.clevelandclinic.org/-/scassets/images/org/health/articles/24000-hurler-syndrome)
Hurler syndrome is the most severe form of mucopolysaccharidosis type 1 (MPS 1). It’s an autosomal recessive condition.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
MPS 1 is a condition where your body doesn't have enough enzymes to break down sugar molecules (glycosaminoglycans, formerly called mucopolysaccharides). The condition causes skeletal/joint abnormalities, distinct facial characteristics, issues with cognitive development, heart and lung (respiratory) problems and an enlarged liver and spleen.
Hurler syndrome is a lysosomal storage condition. When your body is unable to break down molecules normally, they accumulate in lysosomes. Lysosomes are the parts of your cell that control molecular waste management (the storage, recycling and digestion of molecules). When there’s too much toxic molecular buildup in your cells, the cells quickly die or don’t function properly, which causes symptoms of the condition.
Life expectancy for children diagnosed with Hurler syndrome is short due to life-threatening symptoms.
There are three types of Mucopolysaccharidosis type I (MPS I), including:
These types fall on a spectrum based on severity. On one side of the spectrum is Hurler syndrome, which is the most common, most severe and has life-threatening complications. On the other side of the spectrum are Hurler-Scheie syndrome (intermediate form) and Scheie syndrome (mild form). Your child's healthcare provider will most likely call the less severe forms of MPS I “attenuated MPS I.”
Advertisement
Hurler-Scheie and Scheie syndromes have symptoms that match Hurler syndrome, but the progression of the condition is slower. You might see symptoms when your child turns six or seven years old with attenuated MPS I, as opposed to seeing symptoms shortly after birth with Hurler syndrome.
When symptoms of attenuated MPS 1 show up, how they impact the person diagnosed with the condition varies greatly. People with attenuated MPS I can have a normal lifespan and children with Hurler syndrome have a short lifespan.
The main difference between the three types of MPS I is that Hurler syndrome causes major developmental delays during early childhood and affects a child’s intelligence significantly. Hurler syndrome also causes your child’s cognitive abilities to decline over time. Attenuated MPS I can affect a child’s intelligence but not at the same rate as Hurler syndrome.
Hurler syndrome can affect any child since it's the result of a genetic mutation that occurs randomly. If you have a history of mucopolysaccharidosis type I (MPS I) in your family, you’re at an increased risk of having a child with Hurler syndrome.
Hurler syndrome affects an estimated 1 in every 100,000 newborns and affects males and females equally. Less severe forms of mucopolysaccharidosis type I (MPS I) affect nearly 1 out of every 500,000 newborns.
Hurler syndrome affects many aspects of your child’s growing body. Symptoms can cause physical characteristics that are unique to the condition, like an enlarged head, cloudy eyes and facial features such as widely spaced eyes, large forehead, a flat nasal bridge and enlarged lips. Physical symptoms can also affect how your child’s bones grow, which could lead to them having a short stature.
The condition also affects your child’s internal organs, like their heart and lungs. Your child might have recurrent ear, sinus and pulmonary infections or eventually need breathing assistance or surgery to repair symptomatic damage to their organs.
Symptoms of Hurler syndrome are life-threatening and treatment can improve your child’s life expectancy with early diagnosis and treatment.
If you plan on becoming pregnant and want to understand your risk of having a child with a hereditary condition like Hurler syndrome, talk to your healthcare provider about genetic testing.
Symptoms of Hurler syndrome range in severity and are unique to each person diagnosed with the condition. Symptoms begin in early childhood and continue through adolescence.
A symptom of Hurler syndrome that sets it apart from other levels of mucopolysaccharidosis type I (MPS 1) is early childhood developmental delays and a progressive decline in how your child can learn and retain information. Mild cases of MPS 1 don’t affect a child’s intelligence.
Advertisement
Symptoms of Hurler syndrome could include:
During a child’s first year, physical symptoms of Hurler syndrome will appear. These characteristics include:
A mutation of the IDUA gene causes Hurler syndrome. The IDUA gene is responsible for creating lysosomal enzymes, which break down waste in cells. When the IDUA gene doesn't create enough enzymes, toxic waste collects in cells, causing them to die or not function properly. When your cells can’t get rid of waste, symptoms of Hurler syndrome occur.
Hurler syndrome is hereditary, which means you can get the condition from your parents. Hereditary conditions aren't the result of something your parent did while pregnant.
Advertisement
Cells form in your parent’s reproductive organs via one fertilized cell from the sperm and one from the egg. The cells divide and copy themselves with half the amount of DNA as the original cell. During this process of cell division, genetic mutations can occur randomly as cells re-type the instruction manual word for word. Any time there's a typo (genetic mutation), the genetic code for part of your DNA is incomplete. As a result, your cells don’t have the instructions they need to form and function properly.
Prenatal screening tests, like amniocentesis or chorionic villus sampling, can diagnose your child with Hurler syndrome while you’re pregnant. Both tests examine whether or not there are any genetic abnormalities within your baby’s DNA.
After your baby is born, their healthcare provider will diagnose Hurler syndrome with a physical examination to look for symptoms of the condition and enzyme activity assays to confirm the diagnosis. They'll also ask if you know of any family members who have mucopolysaccharidosis conditions, since it’s hereditary.
Additional tests, like an X-ray of your child's bones, echocardiogram of their heart, and blood and urine tests might be necessary to confirm the diagnosis.
Advertisement
Treatment for Hurler syndrome focuses on preventing and managing symptoms of the condition:
Other types of treatment options for Hurler syndrome include:
In some cases, people with Hurler syndrome may have complications related to the anesthetic given during surgical procedures. This happens because your child might have symptoms that cause breathing difficulties and their provider could have trouble securing an IV if they have tight muscles, joints or tissues (contractures).
The timing of treatment is important for it to be effective, especially for hematopoietic stem cell transplantation (HSCT) and enzyme replacement therapy (ERT). If symptoms relating to cognitive development are present, it might be too late for the treatment to work at its full potential.
Before your child begins treatment, talk with their healthcare provider about possible side effects or complications that could arise.
Prognosis is poor for children diagnosed with Hurler syndrome. It’s common for children diagnosed with Hurler syndrome to have a short life expectancy of about 10 years due to the severe symptoms of the condition affecting their heart and lungs. Early diagnosis and treatment can prolong their life with hematopoietic stem cell transplantation (HSCT) and enzyme replacement therapy (ERT).
Children diagnosed with an intermediate or mild form of mucopolysaccharidosis type I (MPS 1) normally live into their early 20s and 30s with treatment. Early death is often from respiratory failure.
If your child’s diagnosis is less severe and treatment begins early, a normal lifespan is possible.
There's no known cure for Hurler syndrome, but treatment prolongs life expectancy and alleviates dangerous symptoms of the condition.
You can’t prevent Hurler syndrome because it’s the result of an inherited genetic mutation. To understand your risk of having a child with a genetic condition, talk to your healthcare provider about genetic testing if you plan on becoming pregnant.
If you notice your child has symptoms of Hurler syndrome, especially if they miss developmental milestones or have trouble seeing or hearing, contact their healthcare provider for an examination.
Visit the emergency room or call 911 immediately if your child has difficulty breathing, an irregular heartbeat or if they pass out regularly (symptoms of cardiomyopathy).
Hurler syndrome and Hunter syndrome are both lysosomal storage conditions that affect the function of lysosomes, which are parts of your cell that control the waste management of molecules.
Hurler syndrome is a severe form of mucopolysaccharidosis type I (MPS I), where your body doesn’t have enough alpha-L-iduronidase enzymes.
Hunter syndrome is less severe than Hurler syndrome. The condition is a form of mucopolysaccharidosis type II (MPS II), where your body doesn’t have enough iduronate-2-sulfatase (I2S) enzymes.
A Hurler syndrome diagnosis can be devastating for your family, especially hearing that your child might have a short life expectancy. During this difficult time, work closely with your child’s healthcare providers to learn more about their diagnosis and treatment options that could help them live longer. It’s also important to surround yourself with support, like your family and friends, along with healthcare providers who can offer emotional comfort during your child’s diagnosis.


Sign up for our Health Essentials emails for expert guidance on nutrition, fitness, sleep, skin care and more.
Learn more about the Health Library and our editorial process.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Do certain health conditions seem to run in your family? Are you ready to find out if you’re at risk? Cleveland Clinic’s genetics team can help.
