Cardiac amyloidosis is a buildup of abnormal proteins in your heart. These proteins thicken your heart muscle, preventing it from working as it should. This ultimately leads to arrhythmias and heart failure. Treatments include medications to slow protein buildup and manage your symptoms. Early diagnosis and treatment improve your outlook.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
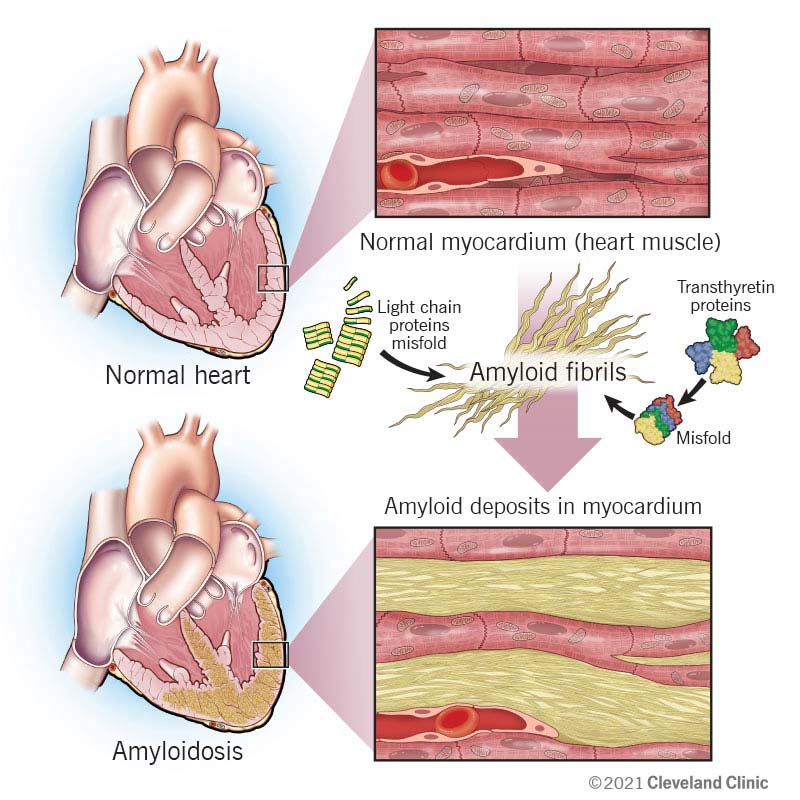
Image content: This image is available to view online.
View image online (https://my.clevelandclinic.org/-/scassets/images/org/health/articles/22598-cardiac-amyloidosis)
Cardiac amyloidosis is a buildup of misshapen proteins in your heart. It’s a leading cause of restrictive cardiomyopathy.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Abnormal proteins build up in your heart muscle. They make the walls of your heart thicken. These changes force your heart to work harder to pump enough blood through your body. The extra effort can weaken and damage your heart. It eventually can lead to heart failure.
If you have cardiac amyloidosis, there’s a good chance you have amyloid buildup in other areas of your body, too. But buildups in your heart tend to cause the most severe health problems. The good news is that recent therapies have improved outcomes for people with this condition.
The two most common types of amyloidosis that can affect your heart are:
Cardiac amyloidosis is a condition that gets progressively worse. It can lead to heart failure symptoms that become more severe with time. They include:
Amyloid can build up elsewhere in your body. So, you may have other symptoms, like peripheral neuropathy (tingling or numbness in your hands or feet) or dysautonomia (lightheadedness, dizziness, low blood pressure with changes in position).
Advertisement
The cause depends on the type of amyloidosis you have. Bone marrow disorders cause AL amyloidosis. A change in the transthyretin (TTR) gene or aging-related changes cause ATTR.
Cardiac amyloidosis changes your heart’s structure, and it can disrupt its electrical system. Complications include:
Healthcare providers diagnose cardiac amyloidosis through various tests. Cardiac amyloidosis is part of a systemic (whole-body) disease. So, providers look at the big picture. In addition to zooming in on your heart, they step back and look for symptoms elsewhere.
Your provider may first suspect this condition based on your symptoms, medical history and physical exam findings. They’ll then use several tests to confirm it or rule it out.
Tests may include:
Your healthcare provider will look for “red flags” that point to cardiac amyloidosis. In addition to heart failure symptoms, they include:
Treatment depends on the type of amyloidosis. In general, there are three main groups of treatments:
Currently, there’s no cure for cardiac amyloidosis. But early diagnosis and treatment can help limit long-term damage to your heart.
Researchers are currently studying other medications that target the underlying causes of cardiac amyloidosis. Ask your healthcare provider if you can join a clinical trial.
You should see your healthcare provider regularly if you have cardiac amyloidosis. You may need frequent appointments to monitor the condition and get treatment.
Ask your providers which symptoms are warning signs that you need emergency care. Share this information with your loved ones so they can get help for you if needed.
Advertisement
How cardiac amyloidosis will affect you depends on many factors, including:
The outlook (prognosis) is generally worse in light-chain amyloidosis due to how quickly it progresses. ATTR has a slower — but still significant — course. Regardless of the type, cardiac amyloidosis is the leading cause of death in people with amyloidosis.
Your healthcare provider can give you a better idea of what to expect. In general, the sooner you receive treatment, the better your outlook and life expectancy.
Cardiac amyloidosis is a life-changing diagnosis. But with each year that passes, researchers learn more about this disease and how to slow it down. Ask your healthcare provider how you can connect with other people who have this condition. Amyloidosis organizations can offer you resources and communities of shared experience. Such support can be vital as you navigate your path forward.
Advertisement


Sign up for our Health Essentials emails for expert guidance on nutrition, fitness, sleep, skin care and more.
Learn more about the Health Library and our editorial process.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
When your heart needs some help, the cardiology experts at Cleveland Clinic are here for you. We diagnose and treat the full spectrum of cardiovascular diseases.
