Charcot-Marie-Tooth (CMT) disease is a group of conditions that affect how your peripheral nerves work. CMT usually affects muscle control and how you feel your feet and hands. It usually isn’t harmful to your health. But it can affect your quality of life. Certain therapies can help with your symptoms.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Charcot-Marie-Tooth (CMT) disease causes worsening weakness in your feet and hands due to peripheral nerve damage. It can also cause sensory-related symptoms, like numbness.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Muscle weakness usually starts in your feet and lower legs. Over time, it can affect your fingers, hands and arms, too. Most people begin to get symptoms in their teen years or early adulthood. But symptoms can start at any age.
CMT disease is the most common inherited (genetic) neuromuscular disorder. But it’s rare overall. It affects about 1 in 2,500 people. The condition is named after the three doctors who first described it.
The symptoms can feel subtle at first. You might notice a bit more clumsiness or feel like your ankles aren’t quite supporting you. If something feels off, reach out to your healthcare provider.
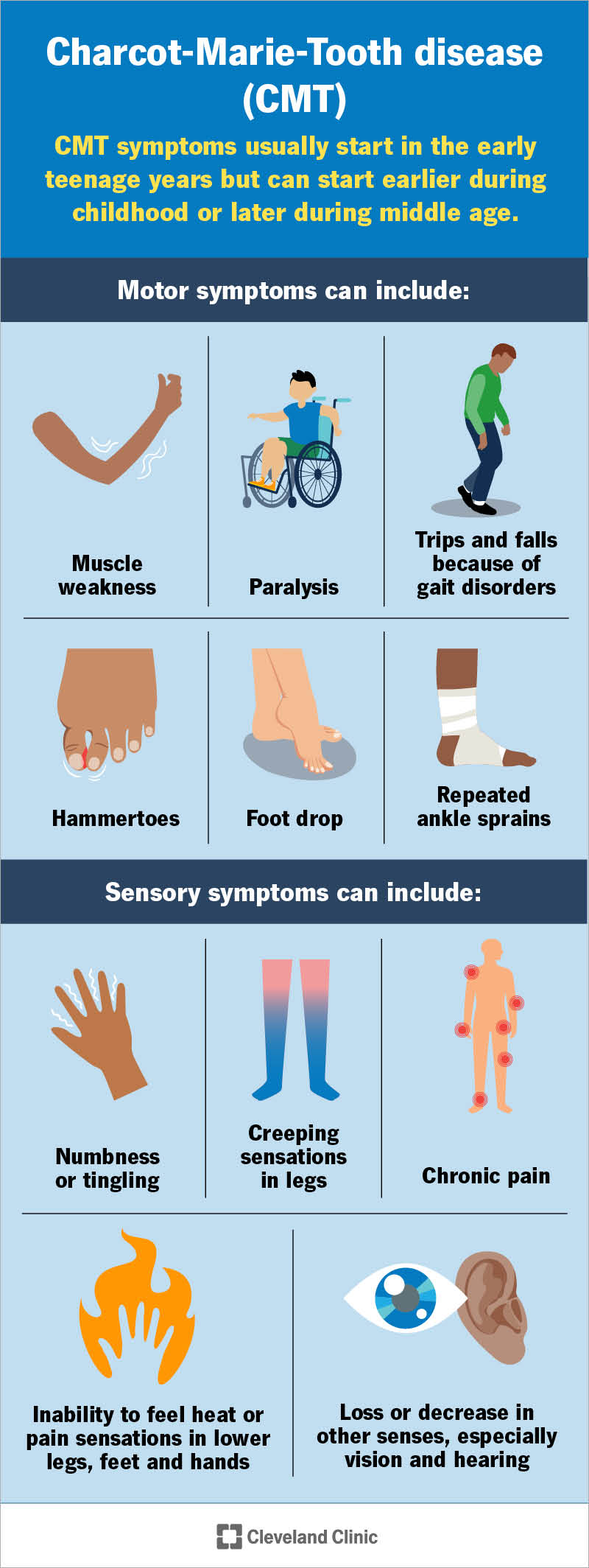
Image content: This image is available to view online.
View image online (https://my.clevelandclinic.org/-/scassets/images/org/health/articles/6009-charcot-marie-tooth-disease)
CMT disease can cause motor (muscle-related) and/or sensory symptoms. Common symptoms include:
These symptoms can range from very mild to severe. Having other conditions that can cause peripheral nerve damage, like diabetes, can make CMT symptoms worse.
Advertisement
Gene changes cause CMT. It can be a change in just one gene or several. Over 100 gene changes are linked to the condition. So, there are many types of it. You inherit the gene change(s) from your biological parent(s). Types of inheritance patterns include:
With CMT, the gene changes affect the cells (neurons) in your peripheral nerves in one of two ways (or both):
The Charcot-Marie-Tooth Association has recorded over 160 subtypes of CMT disease. Most cases fall into four groups.
| Type | Inheritance pattern | Type of nerve damage | Features | Prevalence |
|---|---|---|---|---|
| CMT1 | Autosomal dominant | Myelin loss | Early onset, extremity weakness | About half of CMT cases (most common) |
| CMT2 | Autosomal dominant | Axon damage | Onset in second to third decade of life, extremity weakness | 15% to 30% of cases |
| CMT3 | X-linked | Myelin loss and axon damage | Onset in first to second decade of life; walking issues | 10% to 15% of cases |
| CMT4 | Autosomal recessive | Myelin loss | Progressively severe sensory and muscle symptoms | Less than 10% of cases |
| Type | ||||
| CMT1 | ||||
| Inheritance pattern | ||||
| Autosomal dominant | ||||
| Type of nerve damage | ||||
| Myelin loss | ||||
| Features | ||||
| Early onset, extremity weakness | ||||
| Prevalence | ||||
| About half of CMT cases (most common) | ||||
| CMT2 | ||||
| Inheritance pattern | ||||
| Autosomal dominant | ||||
| Type of nerve damage | ||||
| Axon damage | ||||
| Features | ||||
| Onset in second to third decade of life, extremity weakness | ||||
| Prevalence | ||||
| 15% to 30% of cases | ||||
| CMT3 | ||||
| Inheritance pattern | ||||
| X-linked | ||||
| Type of nerve damage | ||||
| Myelin loss and axon damage | ||||
| Features | ||||
| Onset in first to second decade of life; walking issues | ||||
| Prevalence | ||||
| 10% to 15% of cases | ||||
| CMT4 | ||||
| Inheritance pattern | ||||
| Autosomal recessive | ||||
| Type of nerve damage | ||||
| Myelin loss | ||||
| Features | ||||
| Progressively severe sensory and muscle symptoms | ||||
| Prevalence | ||||
| Less than 10% of cases |
Foot deformities are the main complication of CMT. They tend to get worse over time and can eventually make it impossible to walk without help. Corns, calluses and ulcers can form on your feet, as well.
If you have a loss of sensation in your feet, ulcers and injuries can lead to infection. This is because you may not realize you have them with no feeling of pain.
Your healthcare provider will ask about your medical history and family history. They’ll do physical and neurological exams. You’ll likely need to see a neurologist for further testing.
It may take a combination of tests to get an accurate diagnosis and know the specific type of CMT you have. Tests may include:
There’s no way to cure CMT or treat the condition directly. But therapies and medicines can help manage your symptoms.
Common treatments for CMT include:
Advertisement
Your healthcare provider will suggest various therapies as needed. They’re experts in finding which treatments are most likely to help you based on your specific case.
You’ll have regular follow-up visits with your healthcare provider to check on your symptoms. Reach out to them if you notice any changes, especially ones that disrupt your usual activities or routine.
If you have a fall, get emergency care if there’s any risk of injury to your head, neck or back.
It may be helpful to ask your provider the following questions:
Charcot-Marie-Tooth disease affects each person differently. Most people with CMT develop problems with movement or sensations. But these rarely affect how long you live. With the help of special devices and other kinds of care, it’s possible to do many of the things you love.
Having a condition that gets worse over time can take a toll on your mental health. Reach out to a mental health professional if it’s causing distress.
Advertisement
Living with Charcot-Marie-Tooth disease often means adapting to obstacles that weren’t there before. Aside from the physical symptoms, you may have frustration, grief and uncertainty about what’s next. Despite these challenges, many people with CMT find ways to stay active and engaged with life.
Physical therapy, mobility aids and adjustments can all make a difference. Emotional support — from counseling, support groups or loved ones — matters just as much.
Advertisement


Sign up for our Health Essentials emails for expert guidance on nutrition, fitness, sleep, skin care and more.
Learn more about the Health Library and our editorial process.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Cleveland Clinic’s health articles are based on evidence-backed information and review by medical professionals to ensure accuracy, reliability and up-to-date clinical standards.
Neuropathy, or nerve pain, can make daily life challenging. We can treat the causes of nerve pain and get you back to living life as pain-free as possible.
