What is Marfan Syndrome?
Marfan syndrome (also called Marfan’s syndrome or Marfans syndrome) is a condition that affects the connective tissue. Connective tissue holds the body together and supports many structures throughout the body. Patients with Marfan syndrome, have abnormal connective tissue. As a result, many body systems are affected, including the heart, blood vessels, bones, tendons, cartilage, eyes, nervous system, skin and lungs.
Causes
What causes Marfan syndrome?
Marfan syndrome is caused by a genetic defect. The affected gene is called fibrillin-1 or FBN1. It encodes the structure of fibrillin and the elastic fibers, a major component of connective tissue.
Marfan syndrome is usually an inherited condition and can be passed on to children by just one parent with the condition. If you have Marfan syndrome you have a 50 percent chance of passing it onto your children. The condition is just as common as men as it is in women. The condition is congenital, meaning that it is present at birth, but some patients may not be diagnosed until adolescence or young adulthood.
Marfan syndrome is not always hereditary — 25 percent of patients do not have a parent with the condition and the cause of the genetic defect is unknown.
Who is affected by Marfan syndrome?
Marfan syndrome affects 1 in 10,000 to 20,000 people of all races and ethnic backgrounds.
Symptoms
What are the signs of Marfan syndrome?
Not everyone with Marfan syndrome has the same symptoms and appearance to the same degree. Some patients have few, if any symptoms. In most cases, the disease gets worse with age, and as there are changes in connective tissue, patients notice symptoms.
Physical Appearance
People with Marfan syndrome are often very tall and thin. Their arms, legs, fingers and toes may seem out of proportion and too long for the rest of their body. Their spine may be curved and their breastbone (sternum) may either stick out or be indented. Their joints may be weak and easily become dislocated. Many people with Marfan syndrome have a long, narrow face, and the roof of the mouth may be higher than normal, causing the teeth to be crowded. Not all patients with Marfan syndrome have these characteristics.
Dental and bone problems
Patients with Marfan syndrome often need to have teeth extracted or palate expanders because of a narrow palate. Other problems patients may have are bone problems such as flat feet, hernias and bone dislocations. Other signs of Marfan syndrome can include an abnormal arm span-to-height-ratio and the ability to protrude the thumb past the palm of the hand while making a fist.
Eye problems
More than half of all people with Marfan syndrome have eye problems. These include being nearsighted (trouble seeing objects in the distance), lens subluxation (lens of the eye moves away from its normal position), a difference in the shape of the eye, and other eye problems.
Changes in the heart and blood vessels
The majority (about 90%) of people with Marfan syndrome develop changes in their heart and blood vessels.
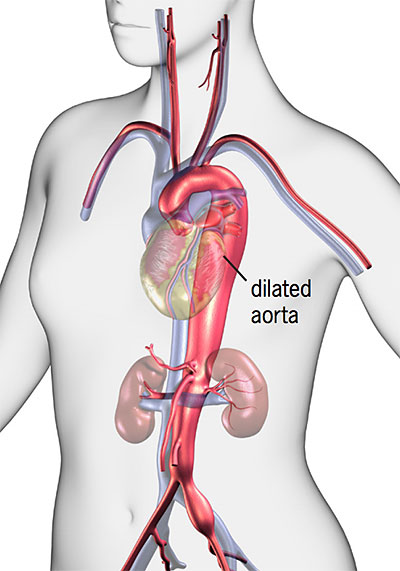
- Blood vessel changes: Marfan syndrome causes the walls of the blood vessels to become weak and dilate (stretch). These changes often affect the aorta, the major artery that carries blood from the heart to the rest of the body. When the walls of the aorta become or stretch, there is a greater risk of aortic aneurysm, aortic dissection or rupture (bursting). Any section of the aorta can dilate or dissect. These conditions can lead to a medical emergency and can be life-threatening. The condition also increases the risk of intracranial bleeding or brain aneurysms, called Berry aneurysms.
- Aortic root dilation: The aortic root is the area where the aorta meets the aortic valve. Marfan syndrome can cause the aortic root to get wider (dilate). This can cause the aortic valve to become stretched and leak. Aortic root dilation is the most common problem with the aorta among patients with Marfan syndrome.
- Heart valve problems: The heart’s valves, especially the mitral valve, can be affected by Marfan syndrome. The valve leaflets become floppy and do not close tightly, allowing blood to leak backwards across the valve (mitral valve prolapse, also called MVP). When MVP progresses, the valve leaks and the condition is called mitral valve regurgitation. Mitral valve prolapse and regurgitation can cause mild to serious valve leakage. Mild valve leaks do not create extra work for the heart, but patients need routine follow-up care. More significant valve leakages cause the heart to work harder and can cause symptoms such as shortness of breath, feeling over-tired, or palpitations (fluttering in the chest). The abnormal flow may cause a heart murmur (abnormal heart sounds heard through a stethoscope). Over time, the heart can get bigger and heart failure can develop.
- Cardiomyopathy: Marfan syndrome can cause the heart muscle to get bigger and weaker over time, causing disease of the heart muscle (cardiomyopathy). This can happen even if the valves are not leaking. Cardiomyopathy can lead to heart failure.
- Arrhythmia (abnormal heart rhythm): Some patients with Marfan syndrome may develop an arrhythmia. This is often related to mitral valve prolapse, but can also happen in patients with a dilated ventricle.
- Lung changes: The changes in lung tissue related to Marfan syndrome increase the risk of asthma, emphysema, chronic obstructive pulmonary disease (COPD), bronchitis, pneumonia and collapsed lung.
- Skin changes: Marfan syndrome causes the skin to become less elastic. This can lead to stretch marks, even without changes in weight.
Aortic Aneurysm
An aortic aneurysm is an abnormal enlargement or bulging of the wall of the aorta. An aneurysm can occur anywhere in the vascular tree.
Once an aneurysm is diagnosed, you may need treatment, depending on the size of the aneurysm. Ruptured aneurysms require emergency surgery to stop the bleeding.
Symptoms of Aortic Aneurysm
- Symptoms of a thoracic aortic aneurysm (affecting upper part of aorta in chest): Pain in the jaw, neck, upper back or chest; coughing, hoarseness or difficulty breathing.
- Symptoms of an abdominal aortic aneurysm (affecting the lower part of the aorta in the abdomen): Pulsating enlargement or tender mass felt by a doctor during a physical exam; pain in the back, abdomen, or groin not relieved with position change or pain medication.
Early diagnosis of an aneurysm is critical to best manage the condition and reduce the risk of rupture.
If you have symptoms of an aneurysm, call your doctor right away.
Aortic Dissection
The aorta has many layers. Aortic dissection is a tear that develops in the inner layer of the aorta, causing blood to flow between the layers. The layers then separate, interrupting the blood flow and possibly causing the arterial wall to burst.
In patients with Marfan syndrome, aortic dissection is a lifethreatening emergency that requires immediate treatment.
Surgical treatment involves repairing or replacing the damaged segment of the aorta.
The goal is to perform surgery BEFORE dissection occurs. This reduces the risk of death and improves the patient’s long-term life expectancy.
The majority of patients who have surgery before a dissection occurs do not need more surgery. However, surgery after an aortic dissection increases the need for future surgeries to repair other sections of the aorta.
Symptoms of Aortic Dissection
- Most common symptom: Severe pain in the chest (front, back or both)
- Less common symptoms: Pain in the abdomen, numbness or weakness in one or both legs, loss of consciousness, or symptoms of a stroke (sudden weakness, numbness, dizziness or loss of balance; sudden vision changes; sudden confusion, difficulty speaking)
If you have symptoms of an aortic dissection, call 911!
Diagnosis
How is Marfan syndrome diagnosed?
Accurate diagnosis of Marfan syndrome requires a multidisciplinary approach because the condition affects many organ systems. Your healthcare team will review your family history and any symptoms you have. You will have a thorough exam that includes your eyes, heart and blood vessels, spine and bones.
You will have tests, such as a chest X-ray, electrocardiogram (ECG) and echocardiogram (an ultrasound of your heart’s valves and chambers). These tests will show changes in the heart and blood vessels, and your heart rhythm.
You may need a transesophageal echo (TEE), magnetic resonance imaging (MRI), or computed tomography (CT) scan if your doctor needs more detailed images or if it is likely you have an aortic dissection.
In addition, a CT or MRI can check for a condition called dural ectasia. This is a bulging in the lining of the spinal column. The condition can cause back pain, but often does not cause any symptoms. Dural ectasia is common in patients with Marfan syndrome but can also be caused by other connective tissue disorders.
A blood test can be used to help diagnose Marfan syndrome. This blood test is highly specialized and is used to detect changes in FBN1, the gene that is responsible for most cases of Marfan syndrome.
Genetic testing should be done in combination with genetic counseling. The FBN1 test is not always straightforward. Blood tests also help diagnose other genetic mutations, such as Loeys-Dietz syndrome, that are similar to Marfan syndrome.
Genetic Testing
What is genetic testing?
Genetic testing is a specialized laboratory test that looks for changes (mutations) in a person’s genetic material (DNA, genes or chromosomes) or in the products that the genes make. Genetic testing is usually done with a blood sample, but other body samples (like cheek cells or skin) are sometimes needed.
The FBN1 test is expensive (approximately $2,000). The cost is often covered by insurance, but you should check with your insurance company about coverage before you have the testing done.
More than 2,000 different mutations have been identified in FBN1; most mutations are unique to an individual family. Once a mutation is found in one family member, the same mutation is likely to appear in other family members who have Marfan syndrome. Sometimes more testing that may involve other family members is needed to confirm Marfan syndrome.
Patients with Loeys-Dietz syndrome may have signs of Marfan syndrome, but are usually not as tall, have elastic skin, have an aortic valve with two (instead of three) leaflets, and a divided or abnormal uvula (the piece of flesh that hangs down at the back of your throat).
Who should have genetic testing?
There are many reasons to consider having genetic testing for Marfan syndrome. You may want to have testing if:
- You have a clinical diagnosis and would like family members to be tested. Genetic testing should begin with the person who has been diagnosed with Marfan syndrome. Once a genetic mutation is identified, other family members can be tested for that mutation at a reduced cost.
- You want to confirm a clinical diagnosis. Genetic testing may confirm a clinical diagnosis of Marfan syndrome. However, genetic testing cannot confirm all cases of Marfan syndrome. If you have a clinical diagnosis without confirmation through genetic testing, it does not mean you do not have Marfan syndrome. It just means that it was not possible to find the genetic reason for the condition with current technology.
- If you had a clinical evaluation but were not diagnosed with Marfan syndrome.
- If Marfan syndrome is one of several diagnoses that are being considered.
- If a family member has a genetic mutation.
Treatments
What treatments are available for patients with Marfan syndrome?
Patients with Marfan syndrome need an individualized treatment plan based on their condition. It is possible that you won’t need treatment, but will need regular follow-up appointments. Treatment may involve medications or surgery.
Medications
Medications are not used to treat Marfan syndrome; however, you may need to take medications to prevent or control complications.
Medications may include:
- Beta-blockers improve the heart’s ability to relax, decreases the forcefulness of the heartbeat and the pressure inside the arteries. This helps prevent or slow the enlargement of the aorta. Betablocker therapy usually begins at an early age. If you cannot take beta-blockers due to asthma or side effects, your doctor may prescribe a calcium channel blocker. Animal experiments show that these drugs can strengthen the aorta.
- Angiotensin receptor blockers (ARBs) are often used to treat patients with high blood pressure as well as heart failure. Recent clinical trials have shown that ARBs help slow the enlargement of the aorta as well as beta blockers do.
Surgery
You may need surgery to prevent an aortic dissection or rupture or to correct a problem with your heart valves. Your doctor may also recommend surgery if you plan to become pregnant.
The need for aortic surgery is based on size of the aorta; expected normal size of the aorta; how fast the aorta is growing; your age, height, and gender; and family history of aortic dissection. Surgery involves a replacement of the dilated portion of the aorta with a graft.
Surgery is not recommended until the aorta is more than 4.7 cm (centimeters) to 5.0 cm (depending on your height) in diameter, or if the aorta is quickly getting wider. Your cardiologist may also calculate your aortic root diameter-to-height ratio. Surgery is recommended if the ratio is greater than 10. During surgery, the dilated section of the aorta is replaced with a graft. Recovery is generally 6 to 8 weeks, including 5 to 10 days in the hospital. Most patients can return to normal activities after they fully recover (ask your doctor about restrictions).
Valve repair or replacement surgery may also be needed if you have a leaky aortic or mitral valve (regurgitation) that leads to shortness of breath, changes in the left ventricle or heart failure.
More information about surgery for patients with Marfan syndrome follows on the next few pages. Please talk to your doctor about any questions you have about surgical treatment options.
Surgical Treatment for Marfan Syndrome
The goal of surgery for patients with Marfan syndrome is to prevent aortic dissection or rupture and treat valve problems. It is recommended that surgery for patients with Marfan syndrome have the procedure done at a major center by a surgeon experienced in treating patients with Marfan syndrome. The combination of experience, early detection, careful follow-up and advanced technology to perform surgery leads to better outcomes.
At Cleveland Clinic, patients with Marfan syndrome or related connective tissue disorders who had surgical treatment had a 99% operative survival rate and an 82% rate for excellent normal expected late survival at 10 years.
Aorta Surgery for Patients with Marfan Syndrome
Two surgical techniques can be used to replace the enlarged area of the aorta with a graft:
- Traditional method: The aorta is replaced with a graft and the aortic valve is replaced with a mechanical valve.
- Valve-sparing modified reimplantation method: The aorta is replaced with a tube graft and the patient’s (native) aortic valve is put back in place. The valve-sparing method is done whenever possible and should be done by an experienced surgeon.
Traditional aorta surgery method
If you have traditional aorta surgery, your surgeon will remove the area of the aorta that has the dissection or aneurysm.
A mechanical valve, attached to the end of an aortic graft, is sewn to the opening (annulus) of the aortic valve.
The coronary arteries are reattached to the aortic graft through small holes cut into the graft. Then, the other end of the graft is sewn to the tissue of the aorta.
Patients who have a mechanical valve need to take anticoagulants (“blood thinners”) for the rest of their lives to prevent blood clots.
Valve-sparing aorta surgery
There are two methods to replace the aorta without replacing the aortic valve:
- Valve sparing re-implantation method
- Valve remodeling method
These techniques can be used in young patients if the aorta is not enlarged and if the aortic valve is not damaged.
Patients who have this surgery do not need to take “blood thinners” for the rest of their lives (unless needed for another condition).
The surgery involves freeing and repairing the aortic valve and replacing the damaged section of the aorta with a synthetic tube graft.
Sutures are placed through the graft and just below the aortic valve, around the left ventricular outflow tract.
A custom-sized dilator device is placed in the left ventricular outflow tract, through the aortic valve. Sutures are tied around the dilator to shape the bottom portion of the aorta graft, similar to the natural aortic root.
The repaired aortic valve is reimplanted and the aortic graft is sewn to the annulus of the repaired valve. The valve is tested to make sure it opens and closes properly.
Small holes are made in the graft for the openings where the coronary arteries are re-attached.
The graft is sewn to the aorta. If the aortic arch needs to be replaced, a separate graft is sewn from the aortic arch to the aortic root graft.
Valve Repair or Replacement Surgery for Patients with Marfan Syndrome
If you have a leaky aortic or mitral valve (regurgitation) that leads to shortness of breath, changes in the left ventricle (left lower chamber of the heart) or heart failure, you may need to have the valve repaired or replaced.
Over the past several years, there have been great advances in the surgical treatment of patients with diseased heart valves. Your doctor will talk to you about available treatments and the best option for you. One of the newest techniques is the modified David’s reimplantation procedure. This procedure involves an aortic graft and keeping the passageway out of the left ventricle and aortic valve in tact. This technique has led to better outcomes.
Cleveland Clinic surgeons are experienced in combining valve surgery with other heart surgeries, such as repair/replacement of more than one valve, bypass surgery, aortic aneurysm surgery or surgery to correct atrial fibrillation (an irregular heartbeat that is common in patients with valve disease). Your surgeon will talk to you about combining procedures if this is the best option for you.
Mechanical Heart Valves
Mechanical heart valves are made of metal or carbon and are designed to perform the functions of your own (native) heart valve. A mechanical valve is very durable, well-tolerated by the body and is designed to last a lifetime.
The bi-leaflet valve is the most common type of mechanical valve and consists of two carbon leaflets in a ring covered with polyester knit fabric.
Life Style Considerations
- Follow-up care: Regular follow-up is important and will include cardiovascular, eye, and skeletal exams, especially while patients are still growing. Your doctors will let you know how often you need to be seen.
- Activity: The amount of activity you can do depends on the extent of the disease and your symptoms. Most people with Marfan syndrome can take part in some type of physical and/or recreational activities. You may need to avoid high-intensity team sports, contact sports, and isometric exercises (such as weight lifting) if you have a dilated aorta. Activities that involve fast movement involving the upper chest and arms while straining should especially be avoided. Ask your cardiologist about the activity guidelines that are right for you.
- Pregnancy: Genetic counseling should be done before pregnancy because Marfan syndrome is an inherited condition. Pregnant women with Marfan syndrome are considered highrisk cases. If the aorta is normal size, the risk of dissection is lower, but still exists. Even a slight enlargement of the aorta causes a greater risk, and the stress of pregnancy can cause the dilation to progress faster than normal. Your doctor may recommend surgery before you become pregnant. Careful follow-up, with frequent blood pressure checks and monthly echocardiograms is required during pregnancy. If there is rapid enlargement or aortic regurgitation, you may need to stay in bed or have surgery. Your doctor will talk to you about the best method of delivery and other details about your care during pregnancy.
- Bacterial endocarditis prevention: People with Marfan syndrome who have had valve or aortic surgery have an increased risk of bacterial endocarditis. This is an infection of the heart valves or tissue caused by bacteria in the blood stream. Talk to your doctor about reducing your risk of bacterial endocarditis by taking antibiotics before having dental or surgical procedures. You can get a card with detailed information about taking preventive antibiotics from the American Heart Association.
References
- Svensson LG, Blackstone EH, Feng J, de Oliveira D, Gillinov AM, Thamilarasan M, Grimm RA, Griffin B, Hammer D, Williams T, Gladish DH, Lytle BW. Are Marfan syndrome and marfanoid patients distinguishable on long-term follow-up? Ann Thorac Surg. 2007 Mar;83(3):1067-74. PMID: 17307461.
- Bhudia SK, Troughton R, Lam BK, Rajeswaran J, Mills WR, Gillinov AM, Griffin BP, Blackstone EH, Lytle BW, Svensson LG. Mitral valve surgery in the adult Marfan syndrome patient. Ann Thorac Surg. 2006 Mar;81(3):843-8. PMID: 16488682.
- Heur M, Costin B, Crowe S, Grimm RA, Moran R, Svensson LG, Traboulsi EI.The value of keratometry and central corneal thickness measurements in the clinical diagnosis of Marfan syndrome. Am J Ophthalmol. 2008 Jun;145(6):997-1001. Epub 2008 Apr 18. PMID:18378212.
- Pearson GD, Devereux R, Loeys B, Maslen C, Milewicz D, Pyeritz R, Ramirez F, Rifkin D, Sakai L, Svensson L, Wessels A, Van Eyk J, Dietz HC; National Heart, Lung, and Blood Institute and National Marfan Foundation Working Group. Report of the National Heart, Lung, and Blood Institute and National Marfan Foundation Working Group on research in Marfan syndrome and related disorders. Circulation. 2008 Aug 12; 118(7):785-91. PMID: 18695204.
- Svensson LG. Aortic valve stenosis and regurgitation: an overview of management. J Cardiovasc Surg (Torino). 2008 Apr;49(2):297-303. PMID: 18431353.
- Svensson LG, Kouchoukos NT, Miller DC, Bavaria JE, Coselli JS, Curi MA, Eggebrecht H, Elefteriades JA, Erbel R, Gleason TG, Lytle BW, Mitchell RS, Nienaber CA, Roselli EE, Safi HJ, Shemin RJ, Sicard GA, Sundt TM 3rd, Szeto WY, Wheatley GH 3rd; Society of Thoracic Surgeons Endovascular Surgery Task Force. Expert consensus document on the treatment of descending thoracic aortic disease using endovascular stent-grafts. Ann Thorac Surg. 2008 Jan;85(1 Suppl):S1-41. PMID: 18083364.
- Svensson LG, Deglurkar I, Ung J, Pettersson G, Gillinov AM, D'Agostino RS, Lytle BW. Aortic valve repair and root preservation by remodeling, reimplantation, and tailoring: technical aspects and early outcome. J Card Surg. 2007 Nov-Dec;22(6):473-9. PMID: 18039206. Svensson LG. The elephant trunk procedure: uses in complex aortic diseases. Curr Opin Cardiol. 2005 Nov;20(6):491-5. Review. PMID: 16234619.
- Svensson LG, Kim KH, Blackstone EH, Alster JM, McCarthy PM, Greenberg RK, Sabik JF, D'Agostino RS, Lytle BW, Cosgrove DM. Elephant trunk procedure: newer indications and uses. Ann Thorac Surg. 2004 Jul;78(1):109-16; discussion 109-16. Review. PMID: 15223413.
- Svensson LG. Sizing for modified David’s reimplantation procedure. Ann Thorac Surg. 2003 Nov;76(5):1751-3. PMID: 14602338.
- Svensson LG, Kim KH, Lytle BW, Cosgrove DM. Relationship of aortic cross-sectional area to height ratio and the risk of aortic dissection in patients with bicuspid aortic valves. J Thorac Cardiovasc Surg. 2003 Sep;126(3):892-3. PMID: 14502185.
- Svensson LG. Progress in ascending and aortic arch surgery: minimally invasive surgery, blood conservation, and neurological deficit prevention. Ann Thorac Surg. 2002 Nov;74(5):S1786-8; Discussion S1792-9. PMID: 12440666.
- Svensson LG, Khitin L. Aortic cross-sectional area/height ratio timing of aortic surgery in asymptomatic patients with Marfan syndrome. J Thorac Cardiovasc Surg. 2002 Feb;123(2):360-1. PMID: 11828302.
- Svensson LG, Longoria J, Kimmel WA, Nadolny E. Management of aortic valve disease during aortic surgery. Ann Thorac Surg. 2000 Mar;69(3): 778-83; Discussion 783-4. PMID: 10750761.
- Svensson LG, Labib SB, Eisenhauer AC, Butterly JR. Intimal tear without hematoma: an important variant of aortic dissection that can elude current imaging techniques. Circulation. 1999 Mar 16;99(10):1331-6. PMID: 10077517.
- Lacro RV, Dietz HC, Sleeper LA, Yetman AT, Bradley TJ, Colan SD, Pearson GD, Selamet Tierney ES, Levine JC, Atz AM, Benson DW, Braverman AC, Chen S, De Backer J, Gelb BD, Grossfeld PD, Klein GL, Lai WW, Liou A, Loeys BL, Markham LW, Olson AK, Paridon SM, Pemberton VL, Pierpont ME, Pyeritz RE, Radojewski E, Roman MJ, Sharkey AM, Stylianou MP, Wechsler SB, Young LT, Mahony L; Pediatric Heart Network Investigators. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N Engl J Med. 2014 Nov 27;371(22):2061-71. Epub 2014 Nov 18. PMID:25405392.
- Svensson LG, Blackstone EH, Alsalihi M, Batizy LH, Roselli EE, McCullough R, Vivacqua A, Moran RT, Gillinov AM, Thamilarasan M, Griffin B, Hammer DF, Stewart WJ, Sabik JF. 3rd, Lytle BW. Midterm results of David reimplantation in patients with connective tissue disorder. Ann Thorac Surg. 2013 Feb;95(2):555-62. Epub 2012 Dec 31. PMID:23286971.
- Svensson LG, Al Kindi AH, Vivacqua A, Pettersson GB, Gillinov AM, Mihaljevic T, Roselli EE, Sabik JF 3rd, Griffin B, Hammer DF, Rodriguez L, Williams SJ, Blackstone EH, Lytle BW. Longterm durability of bicuspid aortic valve repair. Ann Thorac Surg. 2014 May;97(5):1539-47; discussion 1548. Epub 2014 Mar 27. PMID:24680032.
- Svensson LG, Batizy LH, Blackstone EH, Gillinov AM, Moon MC, D’Agostino RS, Nadolny EM, Stewart WJ, Griffin BP, Hammer DF, Grimm R, Lytle BW. Results of matching valve and root repair to aortic valve and root pathology. J Thorac Cardiovasc Surg. 2011 Dec;142(6):1491-8.e7. Epub 2011 Jun 17. PMID:21683965.
Patient Services
Support is Available
We understand that learning you have a genetic disorder, such as Marfan syndrome, is concerning. You may be worried about making lifestyle changes, financial concerns, having surgery, needing to have medical follow-up care the rest of your life. Or there may be financial concerns. It may be also concerning to think about risk to future children.
It is important to seek medical care from a physician who has experience in treating Marfan syndrome. Get accurate information. It may also help to seek genetic counseling to help understand the disease and your risk for passing it on to your children.
In addition to the Marfan syndrome resources at Cleveland Clinic (listed below), these national organizations can help provide you with information and support:
- National Marfan Foundation – 800.862.7326
- National Institute of Arthritis and Musculoskeletal and Skin Disease – 877.226.4267
- American Heart Association – 800.242.8721
Marfan Syndrome and Connective Tissue Disorder Clinic
The multidisciplinary team of experts in the Marfan Syndrome and Connective Tissue Disorder Clinic includes cardiologists, pediatric cardiologists, cardiovascular and vascular surgeons, ophthalmologists, orthopedic surgeons and genetic specialists. Our goal is to help patients live longer and improve their quality of life. We provide:
- A thorough evaluation of patients using state-of-the art diagnostic testing
- Ongoing comprehensive care for patients with disease of the aorta, connective tissue disorder, and Marfan syndrome
- Genetic screening for family members of those with genetic disorders, such as Marfan syndrome
- Ongoing research and education to offer patients high quality and innovative therapies
For More Information
- For more information about Marfan syndrome and treatments
- E-mail us using the Contact Us form on the web
- To talk with a nurse about Marfan syndrome and available treatment options, please contact the Heart & Vascular Resource Center Nurse toll-free at 866.289.6911.
- To make an appointment, please call 800.659.7822.
MyConsult® Online Medical Second Opinion
MyConsult Online Second Opinion program connects patients to the expertise of top Cleveland Clinic specialists without the time and expense of travel. Through our secure web platform, patients can submit their detailed health information, medical records and diagnostic test results. The most appropriate Cleveland Clinic expert is assigned to the consultation and will render a detailed second opinion. The report includes commentary about the diagnosis and treatment options or alternatives and recommendations regarding future therapeutic considerations. Patients are also able to send additional questions to the physician who provided the report. Online medical second opinions are available for more than 1,200 medical diagnoses.
Chat with a Heart Nurse
For over 15 years heart, vascular & thoracic resource nurses have been offering assistance to people who have questions or concerns about heart, vascular and thoracic conditions. Nurses are available to help with questions on symptoms, diagnoses, treatment options, Cleveland Clinic services and doctors, etc. The nurses are available weekdays from 8:30 a.m. – 4 p.m., Eastern Time, through phone, secure live chat or email. If you need help, you may contact a nurse.
Why Choose Us?
About the Sydell and Arnold Miller Family Heart & Vascular Institute
The Sydell and Arnold Miller Family Heart, Vascular & Thoracic Institute at Cleveland Clinic is one of the largest cardiovascular specialty groups in the world, providing patients with expert medical management and a full range of therapies.
We combine research, education and clinical practice to provide innovative and scientifically based treatments for cardiovascular disease. Our physicians and scientists are committed to the prevention and cure of cardiovascular disease. This commitment has led to innovative care, better outcomes and improved quality of life for patients with cardiovascular disease.
Resources
Learn More
- Learn more about marfan syndrome by visiting Cleveland Clinic’s Heart, Vascular & Thoracic Institute website
- Watch videos on aorta disease and treatments presented by Cleveland Clinic specialists
- Read posts about marfan syndrome on our daily blog, Health Essentials from Cleveland Clinic
- Sign up for our e-newsletter, and get tips on maintaining a heart healthy lifestyle, recipes and essential health news
